📋 Key Information Summary
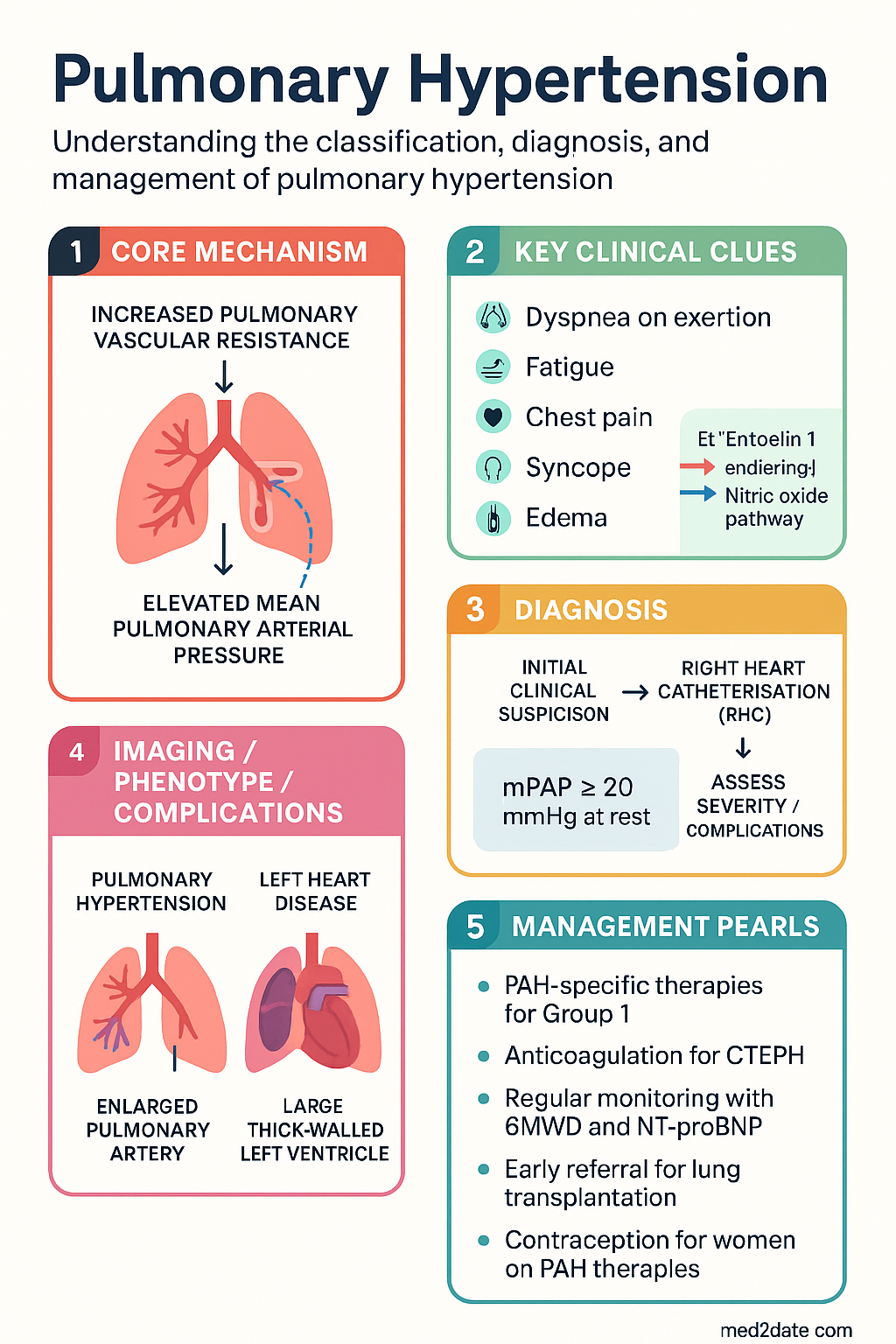
- Pulmonary hypertension (PH) is defined as mean pulmonary arterial pressure (mPAP) >20 mmHg at rest, measured by right heart catheterisation (RHC); pulmonary vascular resistance (PVR) ≥3 WU further characterises pre-capillary disease.
- The WHO Classification divides PH into five groups: Group 1 (PAH), Group 2 (LHD), Group 3 (lung disease), Group 4 (CTEPH), Group 5 (multifactorial); correct classification is critical as therapies are group-specific.
- Group 1 PAH-specific therapies (e.g., Epoprostenol, Sildenafil, Bosentan, Selexipag) are NOT indicated for Groups 2–5 PH and may cause harm if used inappropriately.
- Transthoracic echocardiography (TTE) is the first-line screening tool; RHC is the gold standard for definitive haemodynamic assessment and vasoreactivity testing.
- V/Q scan is mandatory to exclude chronic thromboembolic pulmonary hypertension (CTEPH — Group 4) in all patients with unexplained PH.
- PAH risk stratification uses REVEAL 2.0 or ESC/ERS criteria (low/intermediate/high risk) to guide initial combination therapy decisions.
- Initial dual combination therapy (ERA + PDE5i) is now standard for newly diagnosed PAH; triple therapy is recommended for high-risk patients.
- Epoprostenol (Flolan®, Veletri®) remains first-line for severe/acute vasodilator-responsive PAH; requires continuous IV infusion via central venous access.
- Sildenafil (Revatio®) is PBS-listed (Authority Required) for PAH; Bosentan (Tracleer®) requires PBS Authority approval with hepatology monitoring.
- Pulmonary endarterectomy (PEA) is the treatment of choice for CTEPH (Group 4); balloon pulmonary angioplasty (BPA) is an emerging option for inoperable disease.
- Lung transplantation referral should be discussed early; Australian centres include St Vincent's Hospital Sydney, The Alfred Melbourne, and Prince Charles Hospital Brisbane.
- PH in Aboriginal and Torres Strait Islander peoples is associated with higher prevalence of rheumatic heart disease and chronic lung disease; access to specialist PH centres requires proactive coordination from remote and rural communities.
- All PAH-specific therapies are teratogenic; reliable contraception is mandatory for women of childbearing potential (pregnancy category D — Sildenafil; X — Bosentan, Ambrisentan).
- Serial 6-minute walk distance (6MWD), NT-proBNP, and echocardiographic parameters guide treatment response monitoring every 3–6 months.
- Anticoagulation is recommended for Group 1 PAH (controversial) and mandatory for Group 4 CTEPH with warfarin (target INR 2.0–3.0); DOACs are not recommended for CTEPH.
Introduction & Australian Epidemiology
Pulmonary hypertension (PH) is a haemodynamic condition defined as mean pulmonary arterial pressure (mPAP) >20 mmHg at rest, measured by right heart catheterisation (RHC). Pulmonary arterial hypertension (PAH, Group 1) specifically requires the additional criterion of pulmonary vascular resistance (PVR) ≥3 Wood Units (WU) with a pulmonary arterial wedge pressure (PAWP) ≤15 mmHg.
In Australia, the estimated prevalence of PAH is approximately 15–50 per million population, with an annual incidence of 1–2 per million. Idiopathic PAH (iPAH) is the most common Group 1 subtype, predominantly affecting women aged 30–50 years. Connective tissue disease–associated PAH (CTD-PAH), particularly in systemic sclerosis, accounts for a significant proportion of incident PAH diagnoses.
Group 2 PH (due to left heart disease) is by far the most common cause of PH in Australia, reflecting the high burden of heart failure (~500,000 Australians). Group 3 PH is prevalent in the context of Australia's significant chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD) burden, including dust-related lung diseases. Group 4 CTEPH (chronic thromboembolic pulmonary hypertension) has an estimated prevalence of 5–40 per million, with the diagnosis often delayed due to non-specific symptoms.
Without treatment, Group 1 PAH carries a poor prognosis — median survival historically 2.8 years from diagnosis in iPAH. With modern PAH-specific therapies, survival has improved significantly, with reported 5-year survival rates exceeding 60–70% in contemporary registries. Early diagnosis, correct WHO group classification, and appropriate evidence-based therapy are essential to optimise outcomes.
WHO Classification of Pulmonary Hypertension (Groups 1–5)
The current Nice (6th World Symposium, 2018) WHO classification organises pulmonary hypertension into five clinical groups based on aetiology and pathophysiology. This classification guides investigation, treatment selection, and clinical trial participation.
| WHO Group | Name | Key Subtypes & Examples | Haemodynamic Profile | Therapy Approach |
|---|---|---|---|---|
| Group 1 | Pulmonary Arterial Hypertension (PAH) | 1.1 Idiopathic (iPAH); 1.2 Heritable (BMPR2, ACVRL1, ENG mutations); 1.3 Drug- and toxin-induced (e.g., fenfluramine, dasatinib); 1.4 Associated with CTD (scleroderma, SLE, MCTD); 1.5 HIV infection; 1.6 Portal hypertension (portopulmonary); 1.7 Congenital heart disease (Eisenmenger); 1.8 Schistosomiasis | mPAP >20 mmHg, PVR ≥3 WU, PAWP ≤15 mmHg | PAH-specific therapies: ERAs, PDE5is, sGC stimulators, prostacyclin analogues |
| Group 1′ | Pulmonary Veno-occlusive Disease (PVOD) / Pulmonary Capillary Haemangiomatosis | Often heritable (EIF2AK4 biallelic mutations); rare; can mimic iPAH | Pre- and post-capillary features; low DLCO | PAH drugs used with caution (risk of pulmonary oedema); transplant evaluation early |
| Group 2 | PH Due to Left Heart Disease | 2.1 Heart failure with reduced EF (HFrEF); 2.2 Heart failure with preserved EF (HFpEF); 2.3 Valvular heart disease (mitral/aortic); 2.4 Congenital/acquired inflow/outflow obstruction | PAWP >15 mmHg (isolated post-capillary, IpcPH); ± PVR elevation (combined pre- and post-capillary, CpcPH) | Treat underlying LHD: diuretics, ACEi/ARB, device therapy; NO PAH-specific drugs |
| Group 3 | PH Due to Lung Disease & Hypoxia | 3.1 COPD; 3.2 ILD (IPF, CTD-ILD); 3.3 OHS; 3.4 Developmental lung disorders | Usually post-capillary component; disproportionate PH may warrant PAH Rx discussion | Oxygen therapy, optimise lung disease; PAH drugs not recommended routinely; refer for LTOT if PaO₂ <55 mmHg |
| Group 4 | Chronic Thromboembolic PH (CTEPH) | Organised thrombus with fibrotic remodelling of pulmonary vasculature post-PE | mPAP >20 mmHg, PVR ≥3 WU, organised fibrotic obstruction on CTPA/RHC | Pulmonary endarterectomy (PEA) — curative; BPA for inoperable; Riociguat (Adempas®) PBS-listed |
| Group 5 | PH with Unclear &/or Multifactorial Mechanisms | Haematological (chronic haemolytic anaemia, myeloproliferative); metabolic (GSD, thyroid); systemic (sarcoidosis, Histiocytosis X); others (tumour embolism, CKD/dialysis) | Variable; depends on underlying condition | Treat underlying cause; PAH therapy individualised in specialised centres |
Pathophysiology & Haemodynamics
Normal Pulmonary Haemodynamics
The pulmonary circulation is normally a low-pressure, low-resistance system. Normal values: mPAP 14 ± 3 mmHg, PAWP 6–12 mmHg, PVR 0.3–2.0 WU, cardiac output 5–6 L/min. In PH, chronic elevation of pulmonary arterial pressures leads to right ventricular (RV) pressure overload, RV dilatation, tricuspid regurgitation, and ultimately right heart failure.
Vascular Remodelling in Group 1 PAH
PAH is characterised by a proliferative and anti-apoptotic vascular remodelling process involving all three layers of the pulmonary arterial wall:
- Intimal: Eccentric and concentric intimal fibrosis; plexiform lesions (characteristic of iPAH)
- Medial: Hypertrophy and hyperplasia of vascular smooth muscle cells (VSMCs)
- Adventitial: Fibrosis and inflammatory cell infiltration
- In-situ thrombosis: Microthrombosis in small pulmonary arteries
Key Pathobiological Pathways
Three major molecular pathways are dysregulated in PAH and serve as therapeutic targets:
Haemodynamic Definitions (RHC-Based)
| Parameter | Definition / Threshold | Significance |
|---|---|---|
| Pulmonary Hypertension | mPAP >20 mmHg | Mandatory diagnostic criterion (revised 6th WSPH 2018) |
| Pre-capillary PH (PAH, CTEPH) | mPAP >20 mmHg, PAWP ≤15 mmHg, PVR ≥3 WU | Defines Group 1 (PAH) and Group 4 (CTEPH) |
| Isolated post-capillary PH (IpcPH) | mPAP >20 mmHg, PAWP >15 mmHg, PVR <3 WU | Typical of Group 2 (LHD) |
| Combined pre- and post-capillary PH (CpcPH) | mPAP >20 mmHg, PAWP >15 mmHg, PVR ≥3 WU | Group 2 with significant pulmonary vascular disease; PAH Rx may be discussed at PH MDT |
| Acute vasoreactivity testing (AVT) | IV Epoprostenol or Inhaled Nitric Oxide → ≥10 mmHg fall in mPAP to mPAP ≤40 mmHg, with ↑ or unchanged CO | Positive test → trial of high-dose CCB therapy (limited to ~5% of iPAH) |
Right Ventricular Adaptation and Failure
The RV is a thin-walled, compliant chamber adapted to low-pressure output. In PH, chronic pressure overload causes RV hypertrophy initially (adaptive), followed by dilatation, reduced contractility, and tricuspid regurgitation (maladaptive). RV failure is the most common cause of death in PAH. Biomarkers of RV strain (NT-proBNP, troponin), echocardiographic indices (TAPSE, RV S′, RVFAC, RVGLS), and cardiac MRI-derived RV volumes guide prognosis.
Investigations
Investigation of suspected pulmonary hypertension follows a structured approach: (1) confirm PH on echocardiography, (2) classify by WHO group, (3) assess severity and prognosis. All investigations should be performed at or coordinated through a specialist PH centre.
Essential & Recommended Investigations
Risk Stratification & Severity Scoring
Risk stratification in Group 1 PAH determines treatment goals and escalation. The ESC/ERS 2022 guidelines use a three-strata model (low, intermediate, high risk) based on WHO functional class, 6MWD, NT-proBNP, echocardiography, RHC, and the presence of pericardial effusion.
REVEAL 2.0 Risk Calculator
The REVEAL 2.0 score is widely used in Australian PH centres. It incorporates WHO group, age, sex, renal function (eGFR), BMI, vital signs, 6MWD, NT-proBNP, WHO FC, RA pressure, PVR, pericardial effusion, and DLCO to generate a continuous risk score and category (low, average, moderately high, high, very high). It is validated for reassessment at follow-up visits.
Management of Pulmonary Hypertension
General & Supportive Measures (All Groups)
- Supervised exercise / pulmonary rehabilitation: Improves 6MWD, QoL, and WHO FC; evidence from randomised trials
- Oxygen therapy: Maintain SpO₂ ≥90%; assess need with sleep oximetry; Long-Term Oxygen Therapy (LTOT) if PaO₂ <55 mmHg (PBS-subsidised home oxygen)
- Diuretics: Loop diuretics (Furosemide 20–80 mg PO OD, or Bumetanide 0.5–1 mg PO OD) for RV volume overload and peripheral oedema; monitor electrolytes
- Vaccination: Annual influenza, COVID-19 booster, pneumococcal (Prevenar 13 then Pneumovax 23), pertussis, RSV where eligible
- Psychosocial support: PH diagnosis is associated with significant anxiety and depression; refer to psychology/support groups (Pulmonary Hypertension Association Australasia)
- Avoid: High-altitude travel, pregnancy (mandatory contraception — see Special Populations), strenuous isometric exercise, decongestants (pseudoephedrine), smoking
Anticoagulation in PH
Anticoagulation recommendations vary by WHO group:
- Group 1 PAH: Consider warfarin (INR 2.0–3.0) in iPAH/heritable PAH (observational data only; not RCT-proven). Generally not recommended in CTD-PAH or CHD-PAH due to bleeding risk.
- Group 4 CTEPH: Lifelong anticoagulation is mandatory. Warfarin (INR 2.0–3.0) is preferred; DOACs are NOT recommended due to risk of organised thrombus progression. Riociguat is PBS-listed for inoperable CTEPH.
- Groups 2, 3, 5: Anticoagulation per underlying condition indication (e.g., AF, VTE); no PH-specific benefit.
PAH-Specific Therapy — WHO Group 1 (Drug Detail)
Current Australian guidelines recommend initial dual oral combination therapy (endothelin receptor antagonist + PDE5 inhibitor or sGC stimulator) for newly diagnosed PAH, with escalation to triple therapy including parenteral prostacyclin for intermediate–high risk patients.
Treatment Algorithm — WHO Group 1 PAH (ESC/ERS 2022)
CTEPH (Group 4) — Specific Management
Special Populations
Lung Transplantation in Pulmonary Hypertension
Lung transplantation remains the definitive treatment for selected patients with PAH or CTEPH refractory to maximal medical therapy. Bilateral lung transplantation is the preferred procedure. Heart-lung transplantation is reserved for patients with severe congenital heart disease or inoperable cardiac pathology.
Referral Criteria for Transplant Assessment
- WHO FC III–IV on optimised PAH-specific therapy (including triple therapy with parenteral prostacyclin)
- Declining 6MWD (<330 m) or rising NT-proBNP (>1100 ng/L) despite therapy
- Haemodynamic deterioration: CI <2.0 L/min/m², RA pressure >15 mmHg, mPAP >50% systemic MAP
- Refractory RV failure requiring inotropic support
- Recurrent syncope or severe pericardial effusion
- PVOD / pulmonary capillary haemangiomatosis — early referral recommended
Australian Transplant Centres
| Centre | Location | Capabilities |
|---|---|---|
| St Vincent's Hospital | Sydney, NSW | Bilateral lung transplant; PH programme; PEA |
| The Alfred Hospital | Melbourne, VIC | Bilateral lung transplant; National PEA centre; advanced PH |
| The Prince Charles Hospital | Brisbane, QLD | Bilateral lung transplant; PH programme |
Post-Transplant Considerations
- Immunosuppression: Triple therapy — Tacrolimus + Mycophenolate + Prednisolone (wean to 5 mg OD by 12 months)
- Primary graft dysfunction (PGD) is the leading cause of early mortality
- Chronic lung allograft dysfunction (CLAD) / obliterative bronchiolitis — main cause of late graft loss
- 5-year survival post-lung transplant: approximately 50–60%
Monitoring & Follow-Up
Patients with PAH require lifelong specialist follow-up with a treat-to-target approach. Reassessment at 3–6 month intervals guides treatment escalation decisions.
| Assessment | Frequency | Purpose |
|---|---|---|
| Clinical review (WHO FC, symptoms) | Every 3–6 months | Risk stratification; treatment response |
| NT-proBNP | Every 3–6 months | RV strain biomarker; risk category assignment |
| 6-Minute Walk Test | Every 3–6 months | Functional capacity; prognostic |
| Echocardiography | Every 6–12 months | RV size/function; TR severity; pericardial effusion |
| RHC (haemodynamics) | Every 12–24 months (or if clinical change) | Definitive haemodynamic assessment; treatment goals |
| LFTs (on Bosentan) | Monthly × 3 months, then 3-monthly | Hepatotoxicity screening |
| Hb (on Bosentan) | Baseline, 1 month, 3-monthly | Anaemia monitoring (Bosentan reduces Hb) |
| Pregnancy test (WCBA) | Monthly (on ERA/X-category drugs) | Teratogenicity prevention |
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61(1):2200879. doi:10.1183/13993003.00879-2022
- 2. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. doi:10.1183/13993003.01913-2018
- 3. Galiè N, Barberà JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834–844. doi:10.1056/NEJMoa1413687 (AMBITION trial)
- 4. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. doi:10.1056/NEJMoa1213917 (SERAPHIN trial)
- 5. Ghofrani HA, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–340. doi:10.1056/NEJMoa1209657 (PATENT-1 trial)
- 6. Ghofrani HA, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319–329. doi:10.1056/NEJMoa1209656 (CHEST-1 trial)
- 7. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–2533. doi:10.1056/NEJMoa1503184 (GRIPHON trial)
- 8. Barst RJ, Ivy DD, Gaitan G, et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naïve children with pulmonary arterial hypertension. Circulation. 2012;125(2):324–334. doi:10.1161/CIRCULATIONAHA.110.016667 (SPRINT trial / STARTS-1)
- 9. Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL Risk Score Calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest. 2019;156(2):323–337. doi:10.1016/j.chest.2019.02.004
- 10. Australian Institute of Health and Welfare (AIHW). Rheumatic heart disease and acute rheumatic fever in Australia, 2017–2021. Cat. no. CVD 94. Canberra: AIHW; 2023.
- 11. RHDAustralia (a program of Menzies School of Health Research). RHD in Australia — an overview. Darwin: RHDAustralia; 2023. Available from: rhdaustralia.edu.au
- 12. Keogh AM, McNeil KD, Williams T, et al. Pulmonary hypertension in Australia and New Zealand — the ANZPHTS Registry. Intern Med J. 2009;39(3):168–175. doi:10.1111/j.1445-5994.2008.01784.x
- 13. McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65(18):1976–1997. doi:10.1016/j.jacc.2015.03.540
- 14. Australian Commission on Safety and Quality in Health Care (ACSQHC). National Safety and Quality Health Service Standards. 2nd ed. Sydney: ACSQHC; 2021.
- 15. Moe GW, Howlett J, Januzzi JL, et al. N-terminal pro-B-type natriuretic peptide testing improves the management of patients with suspected acute heart failure. Circulation. 2007;115(24):3103–3110. (Referenced for NT-proBNP use in RV failure)
- 16. The Royal Australian College of General Practitioners (RACGP). Red Book — Guidelines for preventive activities in general practice. 10th ed. East Melbourne: RACGP; 2023. (Vaccination and chronic disease screening guidelines)