📋 Key Information Summary
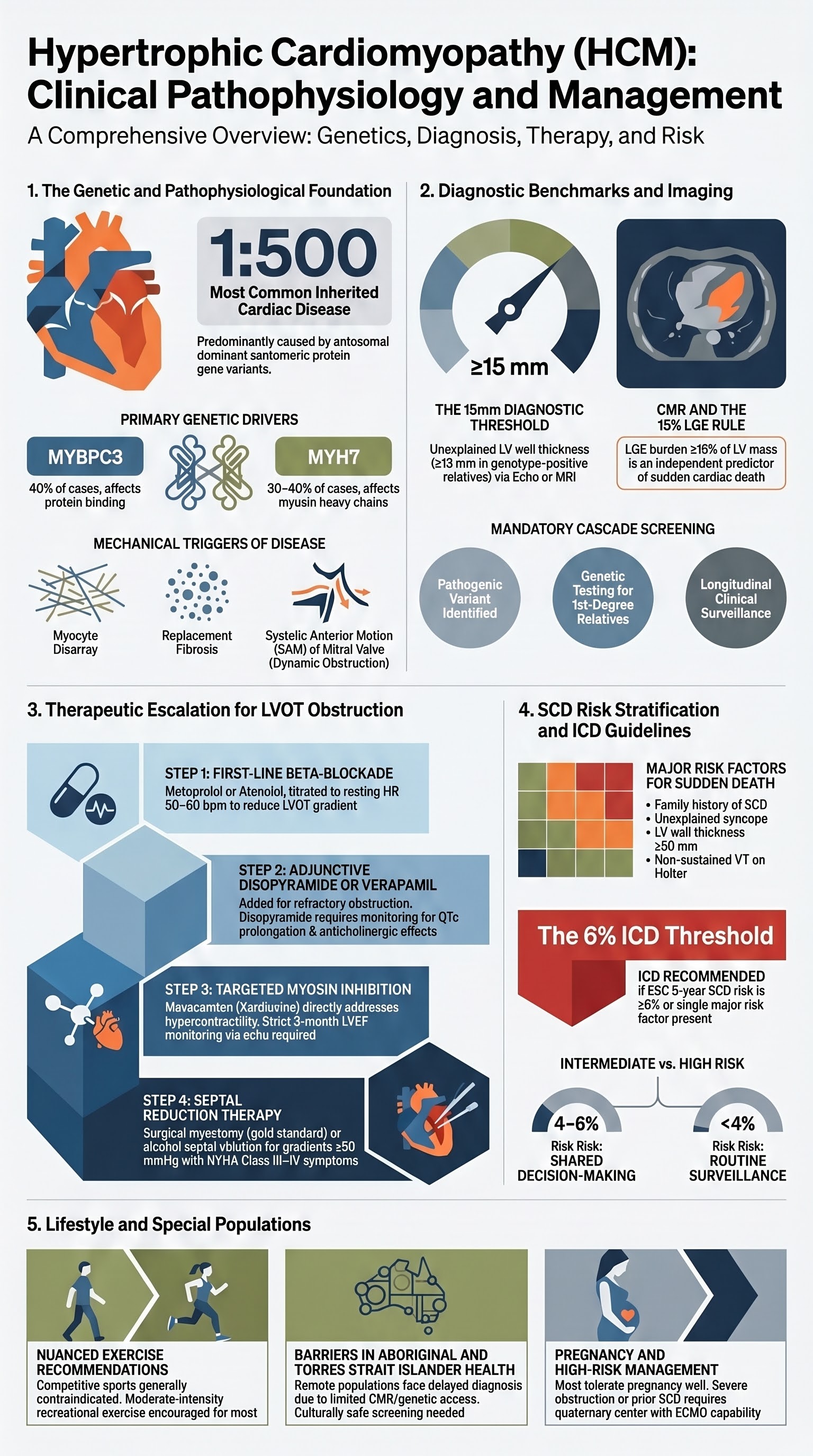
- Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease (prevalence ~1:500), caused predominantly by sarcomeric protein gene variants with autosomal dominant inheritance and variable penetrance.
- Diagnosis requires demonstration of unexplained left ventricular hypertrophy (wall thickness ≥15 mm, or ≥13 mm in genotype-positive relatives) on echocardiography or cardiac MRI, after exclusion of alternative causes.
- Cardiac MRI (CMR) with late gadolinium enhancement (LGE) is essential for risk stratification — LGE burden ≥15% of LV mass is an independent predictor of sudden cardiac death (SCD).
- Genetic testing with a comprehensive HCM panel should be offered to all patients with confirmed HCM; cascade screening of first-degree relatives is mandatory once a pathogenic variant is identified.
- Left ventricular outflow tract (LVOT) obstruction (resting gradient ≥30 mmHg or provoked ≥50 mmHg) drives symptoms and is the primary therapeutic target.
- First-line medical therapy for obstructive HCM is beta-blockers (metoprolol, atenolol); disopyramide (PBS Authority Required) is added as adjunctive therapy for refractory obstruction.
- Mavacamten (Kardiozine®), a first-in-class cardiac myosin inhibitor, is PBS-listed (Authority Required) for symptomatic obstructive HCM refractory to conventional therapy — requires regular echocardiographic monitoring of LVEF.
- Septal reduction therapy (surgical myectomy or alcohol septal ablation) is indicated when maximal medical therapy fails to control symptoms (NYHA Class III–IV).
- SCD risk stratification uses the AHA/ESC risk calculator integrating family history of SCD, unexplained syncope, maximal LV wall thickness ≥30 mm, LVOT gradient, LGE on CMR, and non-sustained VT on Holter.
- ICD implantation is recommended for high-risk patients (5-year SCD risk ≥6% or ≥1 major risk factor) and should be considered for intermediate-risk patients after shared decision-making.
- Competitive sport is generally contraindicated for patients with HCM per AHA/ACC/ESC guidelines; moderate-intensity recreational exercise is permissible with individualised assessment.
- Pregnancy in HCM requires multidisciplinary planning; most women with non-obstructive HCM tolerate pregnancy well, but those with significant obstruction or prior SCD require close cardiology and obstetric co-management.
- Aboriginal and Torres Strait Islander peoples face delayed diagnosis due to geographic barriers and limited access to advanced cardiac imaging; culturally appropriate screening programmes are essential.
Introduction & Australian Epidemiology
Hypertrophic cardiomyopathy (HCM) is a primary cardiac disorder characterised by unexplained myocardial hypertrophy, typically involving the interventricular septum, with a non-dilated, hyperdynamic left ventricle. It is the most common inherited cardiovascular disease, with an estimated prevalence of 1 in 500 in the general population based on echocardiographic screening studies. Genetic studies suggest the true prevalence may be higher, as many carriers of pathogenic sarcomeric variants remain undiagnosed due to incomplete and age-dependent penetrance.
In Australia, HCM is estimated to affect approximately 50,000 individuals, though a significant proportion remain undiagnosed. The condition is the most common cause of sudden cardiac death (SCD) in young people and competitive athletes, accounting for up to one-third of exercise-related SCD in individuals aged <35 years. Australian data from the National Coronial Information System and state-based registries confirm HCM as a leading aetiology in autopsy-positive sudden cardiac arrest in the young.
The natural history of HCM is highly variable. Many patients remain asymptomatic or mildly symptomatic throughout life, while a subset develops progressive heart failure due to diastolic dysfunction, LVOT obstruction, or the development of an end-stage hypokinetic ("burnt-out") phase. Contemporary management has transformed HCM from a disease associated with annual mortality rates of 1–2% (historical data) to a condition with near-normal life expectancy when appropriately managed in specialised centres.
The Australian Institute of Health and Welfare (AIHW) reports that cardiovascular disease remains the leading cause of death in Australia, with cardiomyopathies contributing significantly to premature cardiovascular mortality. The Cardiac Society of Australia and New Zealand (CSANZ) has published position statements on HCM management, and Australian centres contribute actively to international HCM registries including the SHaRe (Sarcomeric Human Cardiomyopathy Registry) and ON-HCM registries.

Pathophysiology
HCM arises from mutations in genes encoding sarcomeric proteins, leading to a complex pathophysiology involving myocyte disarray, myocardial fibrosis, and microvascular dysfunction.
Genetic Basis
Approximately 60% of HCM cases have an identifiable pathogenic or likely pathogenic variant in one of >15 genes encoding sarcomeric proteins. The two most commonly implicated genes account for the majority of genotype-positive cases:
| Gene | Protein | Frequency | Clinical Features |
|---|---|---|---|
| MYBPC3 | Myosin-binding protein C | ~40% of genotype-positive | Later onset, variable penetrance, often frameshift/truncating variants |
| MYH7 | β-myosin heavy chain | ~30–40% | Earlier onset, more severe phenotype, higher penetrance |
| TNNT2 | Cardiac troponin T | ~5% | May have mild hypertrophy but high SCD risk |
| TNNI3 | Cardiac troponin I | ~5% | Variable severity, may cause restrictive physiology |
| TPM1 | α-tropomyosin | ~2–3% | Mild-to-moderate hypertrophy |
| ACTC1 | Cardiac actin | <1% | Rare, mid-ventricular pattern |
Mechanisms of Disease
The fundamental sarcomeric defect produces hypercontractility and impaired relaxation through several mechanisms:
- Myocyte disarray: Disorganised myofibre architecture creates substrate for re-entrant arrhythmias, the primary mechanism of SCD.
- Myocardial fibrosis: Replacement fibrosis (detectable as LGE on CMR) and interstitial fibrosis (detected by T1 mapping/native T1 values) increase myocardial stiffness and arrhythmic substrate.
- Microvascular dysfunction: Reduced coronary flow reserve leads to subendocardial ischaemia, contributing to fibrosis and symptom burden.
- Diastolic dysfunction: Impaired relaxation combined with increased myocardial mass elevates filling pressures, causing exertional dyspnoea and exercise intolerance.
- LVOT obstruction (SAM mechanism): Systolic anterior motion (SAM) of the mitral valve leaflets, caused by the Venturi effect in a narrowed LVOT, creates dynamic obstruction and mitral regurgitation.
Phenotypic Spectrum
HCM demonstrates remarkable phenotypic heterogeneity, even within families carrying the same variant. Disease expression is influenced by genetic modifiers, environmental factors, and comorbidities. Approximately 40% of patients have no identifiable sarcomeric variant (genotype-negative HCM), suggesting additional genetic or non-genetic aetiologies including metabolic/infiltrative storage disorders (Fabry disease, amyloidosis, Danon disease, PRKAG2 syndrome) that must be actively excluded.
Diagnosis & Genetic Testing
Diagnostic Criteria
HCM is diagnosed when imaging demonstrates unexplained left ventricular hypertrophy in the absence of loading conditions that could account for the hypertrophy (e.g., hypertension, aortic stenosis, athlete's heart). The ESC 2023 and AHA/ACC 2024 guidelines define the following criteria:
Echocardiographic Features
Transthoracic echocardiography (TTE) remains the first-line diagnostic investigation. Key features include:
- Asymmetric septal hypertrophy: Interventricular septum disproportionately thickened relative to the posterior wall (ratio typically >1.3:1); most common pattern (~70%).
- Concentric hypertrophy: Uniform wall thickening (~10–15% of cases).
- Apical HCM: Hypertrophy predominantly involving the apex (~10–15%); may be missed on standard parasternal views — requires apical 4-chamber and contrast-enhanced imaging.
- Mid-ventricular obstruction: Obstruction at mid-cavitary level with apical aneurysm formation; higher risk of apical thrombus and SCD.
- SAM of the mitral valve: Systolic anterior motion of the mitral valve leaflet(s) causing LVOT obstruction and a posteriorly directed mitral regurgitation jet.
- LVOT gradient assessment: Resting gradient ≥30 mmHg defines obstruction; provoked gradient ≥50 mmHg with Valsalva manoeuvre, standing, exercise, or post-PVC is clinically significant.
- Diastolic dysfunction: Impaired relaxation (grade I) progressing to restrictive filling (grade III) in advanced disease.
Cardiac MRI (CMR)
CMR with late gadolinium enhancement (LGE) has become an essential investigation in HCM management. Indications include:
- Inconclusive echocardiographic findings or suboptimal acoustic windows.
- Quantification of LV mass, wall thickness (particularly apical segments), and volumes.
- LGE assessment for fibrosis: Present in ~60% of HCM patients. LGE quantified as ≥15% of LV mass is an independent predictor of SCD and ventricular arrhythmias.
- T1 mapping and extracellular volume (ECV) fraction for detection of diffuse interstitial fibrosis.
- Identification of apical aneurysms, mid-ventricular obstruction, and RV involvement.
- Differentiation from athlete's heart, hypertensive heart disease, and infiltrative cardiomyopathies.
Genetic Counselling & Cascade Screening
Genetic counselling is an essential component of HCM evaluation and should be provided both pre- and post-testing:
- Pre-test counselling: Discuss implications of positive, negative, and variant of uncertain significance (VUS) results. Address insurance and employment implications (Australian moratorium on genetic discrimination in life insurance expired March 2024; discuss current protections).
- Post-test counselling: Interpretation of results, family planning implications, and cascade screening recommendation.
- Cascade screening: When a pathogenic or likely pathogenic variant is identified, ALL first-degree relatives should be offered genetic testing. Genotype-positive relatives require longitudinal clinical surveillance (echocardiography and ECG every 1–2 years from age 10–12 years, or earlier if high-risk variant).
- Genotype-negative relatives: Can generally be discharged from surveillance, though expert review is recommended before discontinuing follow-up.
- Genotype-negative/phenotype-positive patients: ~40% of HCM patients; clinical surveillance continues based on phenotype regardless of genetic results.
Obstruction Management
Approximately 70% of HCM patients demonstrate LVOT obstruction at rest or with provocation. Symptom management is centred on reducing the LVOT gradient, improving diastolic filling, and controlling heart rate. Treatment is indicated for patients with an LVOT gradient ≥30 mmHg at rest or ≥50 mmHg with provocation who have symptoms attributable to obstruction (dyspnoea, chest pain, presyncope, syncope).
Medical Therapy
Septal Reduction Therapies
Indicated for patients with severe, drug-refractory LVOT obstruction (gradient ≥50 mmHg at rest or provocation) with persistent NYHA Class III–IV symptoms despite maximal tolerated medical therapy. Referral to a specialist HCM centre is mandatory.
- Gold standard septal reduction therapy
- Open-heart surgery: resection of hypertrophied basal septal muscle (10–15 g) to widen the LVOT
- Relief of obstruction in >95% of patients
- Operative mortality <1% in experienced centres
- Concomitant mitral valve repair/replacement if significant structural MR
- Available at major Australian cardiac surgical centres (Royal Melbourne, Royal Prince Alfred, The Alfred, Monash Health, Flinders Medical Centre)
- Consider extended myectomy (mid-ventricular obstruction) or apical myectomy for non-classic anatomy
- Catheter-based alternative — injection of 1–3 mL absolute ethanol into the septal perforator artery
- Creates a localised myocardial infarction to thin the basal septum
- Gradient reduction comparable to myectomy at experienced centres
- Less invasive but higher rate of permanent pacemaker implantation (10–20% vs 3–5% for surgery)
- Preferred in patients with significant surgical comorbidities or advanced age
- Requires suitable septal perforator anatomy (assessed on coronary angiography)
- Available at interventional cardiology centres in Australian capital cities
Therapy Escalation Pathway
Sudden Death Risk Stratification
Sudden cardiac death (SCD) in HCM is predominantly caused by ventricular tachyarrhythmias arising from myocardial disarray and fibrosis. Risk stratification is a critical component of HCM management, particularly in younger patients, and guides ICD decision-making.
Major Risk Factors
| Risk Factor | Definition | SCD Risk Impact |
|---|---|---|
| Family history of SCD | ≥1 first-degree relative with HCM-related SCD, survived cardiac arrest, or appropriate ICD discharge <40 years | Major risk factor — strongest predictor in young patients |
| Unexplained syncope | ≥1 episode of unexplained syncope within the preceding 6 months (or recurrent, unexplained) | Significant risk factor, particularly if recent or exertional |
| Maximal LV wall thickness ≥30 mm | Greatest wall thickness on any imaging modality | Independent predictor — rare but high-risk finding |
| LVOT obstruction | Resting gradient ≥30 mmHg | Modestly elevated SCD risk; also associated with heart failure progression |
| Non-sustained VT on Holter | ≥3 consecutive ventricular beats at ≥120 bpm on 24–48 hour Holter | Moderate risk factor; sensitivity low but specificity meaningful |
| LGE on CMR | Late gadolinium enhancement ≥15% of LV mass (quantitative) | Independent predictor of SCD and ventricular arrhythmias; increasingly considered a major risk factor |
| Apical aneurysm | Discrete thin-walled dyskinetic/akinetic apical segment | Associated with mid-ventricular obstruction, scar, and VT/VF — high-risk feature |
| Systolic dysfunction (LVEF <50%) | End-stage "burnt-out" phase | Elevated SCD and heart failure mortality risk |
ESC HCM Risk-SCD Calculator
The ESC 2023 guidelines endorse the HCM Risk-SCD calculator (developed by the ESC HCM Outcome Investigators) to estimate 5-year SCD probability. The calculator integrates age, maximal LV wall thickness, LA diameter, LVOT gradient, family history of SCD, unexplained syncope, and NSVT.
ICD Indications in HCM
| Indication | Class | Details |
|---|---|---|
| Survived cardiac arrest or sustained VT | Class I | Secondary prevention — ICD recommended regardless of risk factors |
| ≥1 major SCD risk factor | Class I | Family SCD, unexplained syncope, max wall ≥30 mm, LGE ≥15%, apical aneurysm, LVEF <50% |
| ESC Risk-SCD ≥6% | Class I | Calculator-based high risk |
| ESC Risk-SCD 4–6% | Class IIa | ICD should be considered with shared decision-making |
| ESC Risk-SCD <4% without major risk factors | Class III | ICD generally not recommended — risks may outweigh benefits |
Family Screening Protocols
Systematic family screening is a cornerstone of HCM management and SCD prevention:
- Genotype-positive families: Cascade genetic testing for all first-degree relatives. Genotype-positive/phenotype-negative individuals: clinical surveillance with ECG and echocardiography every 1–2 years from age 10–12 years (or 5 years before the earliest diagnosis in the family), with transition to adult HCM services at age 16–18 years.
- Genotype-negative families: If the proband is genotype-negative (no pathogenic variant identified), first-degree relatives should have a single clinical screening (ECG + echocardiography). If normal, they can be discharged from surveillance, though repeat screening at 3–5 year intervals may be considered if clinical suspicion exists.
- Athletes: Pre-participation screening in Australia (Australian Institute of Sport protocols) includes ECG. Further investigation with echocardiography is triggered by abnormal ECG findings.
- Post-mortem genetic testing: When SCD occurs in a suspected HCM case, post-mortem genetic testing (molecular autopsy) should be offered to inform family screening. Available through the Australian National Coronial Information System and specialist forensic pathology services.
Exercise & Lifestyle
Exercise Recommendations
Exercise counselling in HCM is complex and must be individualised. Historically, all competitive sport was contraindicated. The 2024 AHA/ACC guidelines introduced a more nuanced approach acknowledging that moderate-intensity recreational exercise is safe and beneficial for most HCM patients.
| Activity Level | Recommendation | Rationale |
|---|---|---|
| Competitive sport (high-intensity) | Generally contraindicated (Class III / Class IIb in selected low-risk patients per AHA 2024) | Exercise-related SCD risk; haemodynamic stress of intense exertion. Individualised shared decision-making permissible for low-risk patients per AHA 2024 update. |
| Moderate-intensity recreational exercise | Encouraged for most patients (Class I) | Improves functional capacity, quality of life, and cardiovascular fitness. Activities: brisk walking, cycling, swimming at comfortable pace. Target 150 min/week. |
| Low-intensity activity | No restrictions | Walking, gentle yoga, daily activities. Encourage as baseline. |
| Isometric/heavy resistance training | Avoid or limit to light weights with proper breathing technique | Valsalva manoeuvre acutely increases LVOT gradient; risk of syncope and arrhythmia. |
Lifestyle Counselling
- Hydration and heat: Avoid dehydration and excessive heat exposure — both reduce preload and can worsen LVOT obstruction and cause hypotension.
- Alcohol: Moderate or avoid alcohol. Acute alcohol consumption has negative inotropic effects but may cause vasodilation and worsen obstruction. Chronic heavy alcohol use may contribute to additive cardiomyopathy.
- Driving: Australian driving restrictions apply: patients with HCM and ICD implantation must notify their state/territory licensing authority. Austroads guidelines recommend cessation of private vehicle driving for 4 weeks post-ICD implantation and 6 months following appropriate ICD therapy. Commercial driving restrictions are more stringent.
- Diving: Scuba diving is generally contraindicated in symptomatic HCM or those with significant obstruction due to risk of syncope underwater and haemodynamic stress.
- Medications to avoid: Vasodilators (nitrates, PDE-5 inhibitors, ACE inhibitors/ARBs) may worsen obstruction. Decongestants containing pseudoephedrine may trigger arrhythmias. Discuss with cardiologist before starting any new medication.
- Endocarditis prophylaxis: NOT routinely recommended for HCM alone (per ESC/AHA guidelines). Only indicated if concurrent valvular disease or prosthetic material.
Pregnancy Management in HCM
Most women with HCM tolerate pregnancy well, particularly those without significant obstruction and preserved LV function. However, pregnancy-related haemodynamic changes (increased blood volume, decreased SVR, increased HR) can exacerbate LVOT obstruction and heart failure symptoms.
Special Populations
Paediatric HCM
Pregnancy
Elderly Patients
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal and Torres Strait Islander Health Considerations
Emerging Therapies
The therapeutic landscape for HCM is rapidly evolving, driven by improved understanding of the molecular pathophysiology. The advent of cardiac myosin inhibitors represents the first disease-modifying therapy for HCM, and multiple pipeline agents are in development.
Cardiac Myosin Inhibitors
Gene Therapy Research
Gene therapy for HCM is in early-phase investigation, targeting the fundamental genetic defect rather than downstream pathophysiology:
- Gene silencing (ASO/siRNA): Antisense oligonucleotides (ASOs) targeting mutant MYBPC3 mRNA are being developed to reduce expression of the dominant-negative protein. Preclinical studies in HCM mouse models show reversal of hypertrophy and fibrosis. Clinical trials are in planning stages.
- Gene replacement therapy: AAV-based delivery of functional MYBPC3 to cardiomyocytes. Significant challenges remain: immune response to AAV vectors, achieving sufficient cardiac tropism, and ensuring sustained expression.
- CRISPR-based approaches: Preclinical research into allele-specific editing of pathogenic MYH7 or MYBPC3 variants. Ethical and delivery challenges are substantial, but long-term potential for curative therapy exists.
- mRNA therapy: Lipid nanoparticle-delivered mRNA encoding functional cardiac proteins — an emerging platform building on COVID-19 vaccine technology. Early preclinical work showing promise.
Other Pipeline Therapies
| Agent | Mechanism | Phase | Target Population |
|---|---|---|---|
| Aficamten (CK-274) | Cardiac myosin inhibitor | Phase III | Obstructive and non-obstructive HCM |
| Bimekizumab-related research | Anti-fibrotic (targeting cardiac fibrosis) | Preclinical | HCM with significant fibrosis |
| N-acetylcysteine (NAC) | Antioxidant/anti-fibrotic | Phase II (small studies) | Non-obstructive HCM with LV fibrosis |
| Eleclazine (GS-6615) | Late sodium current inhibitor | Phase II (discontinued — mixed results) | HCM with arrhythmia burden |
| Ranolazine | Late sodium current inhibitor | Phase II (RHYTHM-HCM) | HCM with ventricular arrhythmias and diastolic dysfunction |
| Perhexiline | Metabolic modulator (CPT-1 inhibitor) | Phase II (Australian-led trials) | Non-obstructive HCM — improves diastolic function by shifting myocardial metabolism from fatty acid to glucose oxidation |
Clinical Trial Opportunities in Australia
Australia is an active contributor to international HCM research, with clinical trial sites at major centres:
- ANZCTR (Australian New Zealand Clinical Trials Registry): Search for current HCM trials at anzctr.org.au.
- Active Australian HCM research centres: Baker Heart and Diabetes Institute (Melbourne), Victor Chang Cardiac Research Institute (Sydney), Royal Melbourne Hospital HCM Programme, Centenary Institute (Sydney), and multiple university-affiliated hospitals.
- Patient registries: The SHaRe Registry (Sarcomeric Human Cardiomyopathy Registry) includes Australian sites and enables genotype–phenotype correlation research. Encourage patients to enrol in registries for research and surveillance.
- Clinical trial access: Patients with refractory symptoms, genotype-positive status, or interest in contributing to research should be referred to their nearest HCM specialist centre for discussion of available trials. Trial eligibility typically requires confirmed HCM diagnosis and may have genotype-specific enrolment criteria.
📚 References
- 1. Ommen SR, Ho CY, Asif IM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2024;149(23):e1239–e1311.
- 2. Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the Management of Cardiomyopathies. Eur Heart J. 2023;44(37):3503–3626.
- 3. Maron BJ, Desai MY, Nishimura RA, et al. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022;79(4):372–389.
- 4. Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396(10261):759–769.
- 5. Braunwald E, Olivotto I, Maron MS, et al. Mavacamten: a first-in-class myosin inhibitor for obstructive hypertrophic cardiomyopathy. Eur Heart J. 2023;44(46):4622–4633.
- 6. O'Mahony C, Jichi F, Ommen SR, et al. International External Validation Study of the 2014 European Society of Cardiology Guidelines on Sudden Cardiac Death Prevention in Hypertrophic Cardiomyopathy (EVIDENCE-HCM). Circulation. 2018;137(10):1015–1023.
- 7. Maron BJ, Udelson JE, Bonow RO, et al. Eligibility and Disqualification Recommendations for Competitive Athletes with Cardiovascular Abnormalities: Task Force 3: Hypertrophic Cardiomyopathy, Arrhythmogenic Right Ventricular Cardiomyopathy and Other Cardiomyopathies, and Myocarditis. Circulation. 2015;132(22):e273–e280.
- 8. Australian Institute of Health and Welfare. Cardiovascular disease in Australia. AIHW. Canberra; 2024.
- 9. Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249–1254.
- 10. Ho CY, Day SM, Ashley EA, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138(14):1387–1398.
- 11. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy: preparticipation screening for athletes and the role of cardiac magnetic resonance imaging. JACC Cardiovasc Imaging. 2009;2(2):170–177.
- 12. Desai MY, Owens A, Geske JB, et al. Dose-Blinded Myosin Inhibition in Patients with Obstructive Hypertrophic Cardiomyopathy Receiving Background Therapy: The VALOR-HCM Randomized Clinical Trial. JAMA Cardiol. 2023;8(7):624–632.
- 13. Ingles J, Burns C, Bagnall RD, et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ Cardiovasc Genet. 2017;10(2):e001620.
- 14. Heart Foundation. Aboriginal and Torres Strait Islander heart health. Heart Foundation Australia. 2023.
- 15. Noaman S, Andrianopoulos N, Brennan A, et al. Trends in cardiovascular disease in Aboriginal and Torres Strait Islander peoples. Med J Aust. 2022;217(S8):S5–S12.