📋 Key Information Summary
- Long QT syndrome (LQTS) is a disorder of cardiac repolarisation characterised by prolongation of the corrected QT interval (QTc) on ECG, predisposing to torsades de pointes (TdP) ventricular tachycardia and sudden cardiac death (SCD).
- Congenital LQTS is caused by pathogenic variants in genes encoding cardiac ion channels; the three most common subtypes — LQTS1 (KCNQ1), LQTS2 (KCNH2), and LQTS3 (SCN5A) — account for >90% of genotype-positive cases.
- Acquired LQTS is most frequently drug-induced (antiarrhythmics, antipsychotics, antibiotics, antiemetics) but may also result from electrolyte disturbances, hypothyroidism, or structural heart disease.
- Diagnosis rests on the Schwartz score incorporating ECG findings, clinical history, and family history; QTc ≥470 ms in males or ≥480 ms in females on a resting 12-lead ECG is highly suggestive.
- Genetic testing is indicated in patients with a Schwartz score ≥3.5, unexplained syncope, or a first-degree relative with confirmed LQTS; available through Australian clinical genetics services (MBS item 73291).
- LQTS1 triggers include swimming and exertion; LQTS2 triggers include auditory stimuli and emotional stress; LQTS3 events often occur at rest or during sleep.
- Non-selective beta-blockers (nadolol, propranolol) are first-line therapy for congenital LQTS; atenolol and metoprolol are less effective and not recommended as primary agents.
- Left cardiac sympathetic denervation (LCSD) is an option for patients with recurrent events on beta-blockers or those intolerant of beta-blockade; performed at select Australian centres.
- ICD implantation is recommended for survivors of cardiac arrest and should be considered for patients with recurrent syncope on maximised beta-blocker therapy.
- All patients must avoid QT-prolonging medications; maintain an updated QT-drug list via crediblemeds.org (QTDrugs database). Pharmacy alert systems and My Health Record annotations are recommended.
- Electrolyte correction — ensure serum potassium ≥4.0 mmol/L and magnesium ≥0.8 mmol/L; hypokalaemia is the single most important modifiable risk factor for TdP.
- Paediatric considerations: LQTS1 and LQTS2 present in childhood; swimming restrictions for LQTS1; beta-blocker doses weight-based; family screening of first-degree relatives is mandatory.
- Aboriginal and Torres Strait Islander patients face barriers to specialist cardiac and genetic services in remote areas; telehealth ECG review and culturally safe cascade screening programmes are essential.
Introduction & Australian Epidemiology
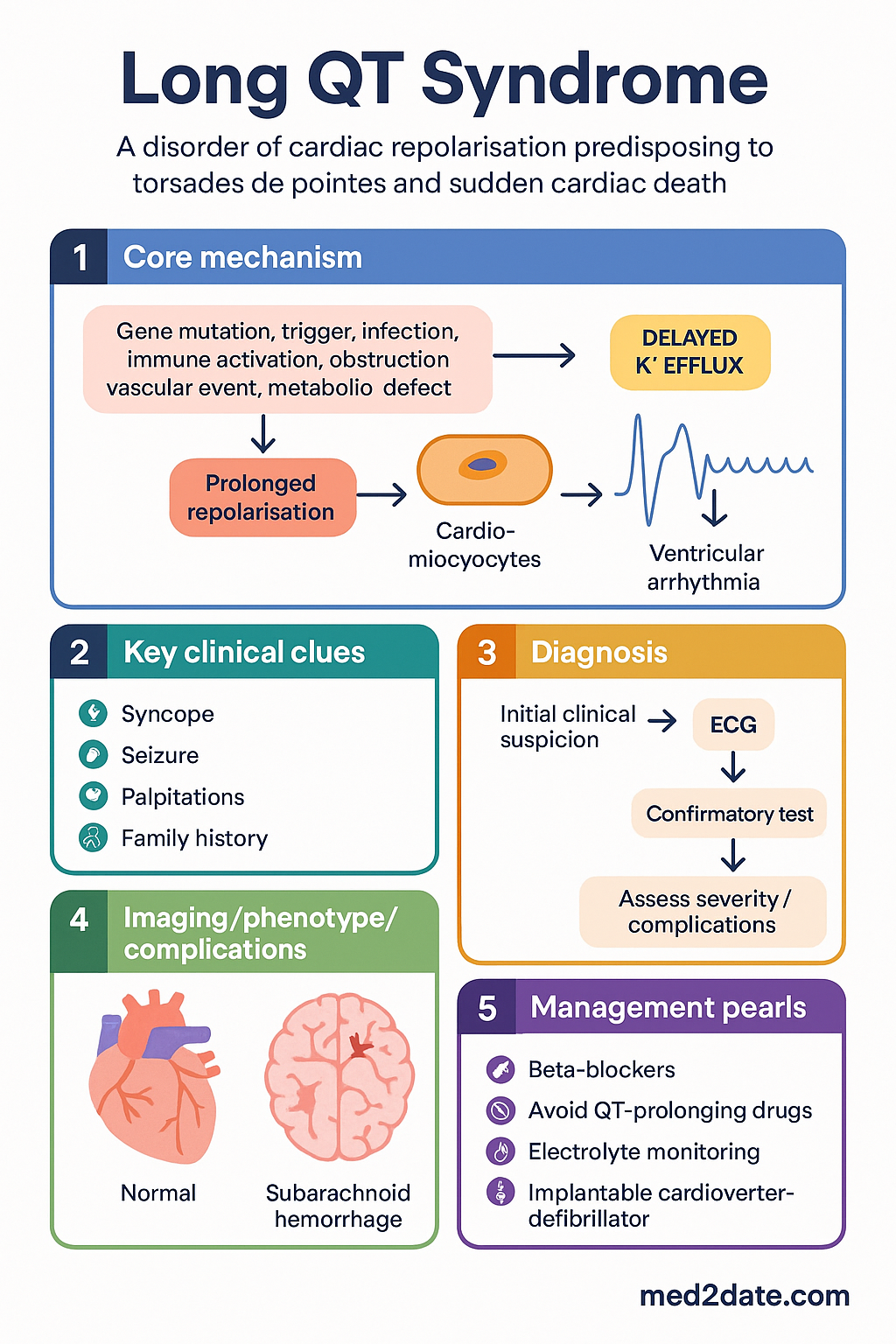
Long QT syndrome (LQTS) is an inherited or acquired channelopathy of cardiac repolarisation that predisposes affected individuals to the polymorphic ventricular tachycardia torsades de pointes (TdP) and sudden cardiac death (SCD). The condition is characterised by prolongation of the QT interval on the surface electrocardiogram (ECG), reflecting delayed repolarisation of ventricular myocytes due to dysfunction of membrane ion channels responsible for the cardiac action potential.
Congenital LQTS is caused by pathogenic variants in genes encoding potassium or sodium channel subunits, inherited in an autosomal dominant pattern (Romano-Ward syndrome) or, rarely, in a recessive pattern associated with sensorineural deafness (Jervell and Lange-Nielsen syndrome). Over 17 genes have been implicated, but three major subtypes — LQTS1 (KCNQ1), LQTS2 (KCNH2), and LQTS3 (SCN5A) — account for more than 90% of genotype-positive cases.
Acquired LQTS is considerably more prevalent and is most commonly iatrogenic, resulting from exposure to medications that block the hERG (IKr) potassium channel. Numerous drug classes are implicated, including Class IA and III antiarrhythmics, certain antibiotics (fluoroquinolones, macrolides), antipsychotics (haloperidol, droperidol, ziprasidone), antiemetics (ondansetron, domperidone), and methadone.
In Australia, the National Coronial Information System (NCIS) and the SADS (Sudden Arrhythmic Death Syndrome) Foundation have highlighted the burden of inherited arrhythmia syndromes. SADS Australia estimates that genetic cardiac conditions cause approximately 500 sudden cardiac deaths per year in Australians under 50, with LQTS being a leading identified cause. The Cardiac Society of Australia and New Zealand (CSANZ) provides expert consensus guidance on the evaluation and management of inherited arrhythmia syndromes.
Key Australian resources include the SADS Foundation Australia, the CSANZ Inherited Arrhythmias Working Group, and the Royal Australasian College of Physicians (RACP) guidelines on genetic evaluation of sudden cardiac death. Genetic testing is available through public clinical genetics services in each state and territory, with MBS rebates under items for genomic sequencing where criteria are met.
Congenital LQTS: Genetics & Types
Congenital LQTS is caused by loss-of-function or gain-of-function variants in genes encoding cardiac ion channel α-subunits or their regulatory proteins. The three major subtypes have distinct genetic aetiologies, clinical triggers, ECG features, and responses to therapy.
| Subtype | Gene | Channel / Current | Prevalence | Typical Triggers | ECG Features |
|---|---|---|---|---|---|
| LQTS1 | KCNQ1 | ↓ IKs (slow delayed rectifier K⁺) | ~40% of genotype+ cases | Exercise (especially swimming), emotional stress, sudden auditory stimuli (alarm clock) | Broad-based T wave; QTc often normalises at rest but prolongs with sympathetic activation |
| LQTS2 | KCNH2 (hERG) | ↓ IKr (rapid delayed rectifier K⁺) | ~30% of genotype+ cases | Emotional stress, startle, post-partum period, auditory stimuli | Low-amplitude bifid or notched T wave in precordial leads |
| LQTS3 | SCN5A | ↑ INa (persistent inward Na⁺ current) | ~10% of genotype+ cases | Rest, sleep, bradycardia, fever | Late-onset peaked/biphasic T wave; long isoelectric ST segment |
Less Common Subtypes
Rare subtypes (LQTS4–LQTS17) account for <5% of genotype-positive cases and include variants in ANK2 (LQTS4), KCNE1 (LQTS5), KCNE2 (LQTS6), KCNJ2 (LQTS7, Andersen-Tawil syndrome), CACNA1C (LQTS8, Timothy syndrome), CAV3 (LQTS9), KCNJ5 (LQTS13), and SCN4B (LQTS10). Many of these overlap with catecholaminergic polymorphic ventricular tachycardia (CPVT) or Brugada syndrome phenotypes.
Jervell and Lange-Nielsen Syndrome
Autosomal recessive inheritance of biallelic KCNQ1 or KCNE1 pathogenic variants causes the Jervell and Lange-Nielsen syndrome (JLNS), characterised by congenital sensorineural deafness and a severe LQTS phenotype with very long QTc intervals and a high risk of cardiac events in early childhood. JLNS prevalence is estimated at 1 in 200,000.
Genetic Testing in Australia
Genetic testing for LQTS is available through Australian clinical genetics services and should be offered to:
- Patients with a Schwartz score ≥3.5 (intermediate or high probability).
- Patients with unexplained syncope, seizure, or cardiac arrest where LQTS is a differential.
- First-degree relatives of genotype-positive probands (cascade testing).
- Cases of sudden unexplained death in the young referred for molecular autopsy.
Testing is performed by next-generation sequencing (NGS) panels targeting the major LQTS genes, with whole-exome or whole-genome sequencing available for complex phenotypes. MBS item 73291 provides a rebate for genomic testing where clinical criteria are met. Results should be interpreted by a clinical geneticist or cardiologist with expertise in inherited arrhythmias, and variant classification follows American College of Medical Genetics and Genomics (ACMG) criteria.
Acquired LQTS: Causes & Drug-Induced
Acquired QT prolongation is far more common than congenital LQTS and is predominantly iatrogenic. Drug-induced QT prolongation results from blockade of the hERG (IKr) potassium channel encoded by KCNH2, though other mechanisms (INa augmentation, ICaL effects) are increasingly recognised. Importantly, a proportion of patients who develop drug-induced TdP harbour clinically silent pathogenic variants in LQTS genes — so-called "latent" or "concealed" congenital LQTS — which unmask under pharmacological or metabolic stress.
High-Risk QT-Prolonging Medications
| Drug Class | Examples (Australian brands) | TdP Risk | PBS Status |
|---|---|---|---|
| Class III antiarrhythmics | Amiodarone (Arycor®), sotalol (Sotacor®), dofetilide | High | PBS General Benefit |
| Class IA antiarrhythmics | Disopyramide (Rythmodan®), procainamide | High | PBS General Benefit |
| Antipsychotics | Haloperidol (Haldol®), droperidol (Droleptan®), ziprasidone (Zeldox®), thioridazine | High | PBS General Benefit |
| Antiemetics | Ondansetron (Zofran®), domperidone (Motilium®) | Moderate | PBS General Benefit |
| Fluoroquinolone antibiotics | Moxifloxacin (Avelox®), ciprofloxacin (Ciproxin®) | Moderate | PBS Restricted Benefit |
| Macrolide antibiotics | Erythromycin, clarithromycin (Klacid®), azithromycin (Zithromax®) | Moderate | PBS General Benefit |
| Antifungals | Fluconazole (Diflucan®), itraconazole (Sporanox®) | Moderate | PBS General Benefit |
| Opioids | Methadone (Physeptone®), levomethadyl | High | PBS Authority Required |
| Antimalarials | Chloroquine, hydroxychloroquine (Plaquenil®) | Moderate | PBS General Benefit |
Non-Drug Causes of Acquired QT Prolongation
- Electrolyte disturbances: Hypokalaemia (K⁺ <3.5 mmol/L), hypomagnesaemia (Mg²⁺ <0.7 mmol/L), hypocalcaemia (Ca²⁺ <2.1 mmol/L).
- Endocrine: Hypothyroidism (bradycardia-related QT prolongation), hypoparathyroidism.
- Nutritional: Anorexia nervosa, liquid protein diets, celiac disease (electrolyte depletion).
- Cardiac: Myocarditis, ischaemic cardiomyopathy, significant bradycardia (sinus node dysfunction, complete heart block).
- Neurological: Subarachnoid haemorrhage, stroke (central sympathetic surge).
- HIV infection: Direct viral myocardial effects and antiretroviral drugs (efavirenz, lopinavir/ritonavir).
Risk Factors for Drug-Induced TdP
The following patient factors increase susceptibility to drug-induced QT prolongation and TdP:
- Female sex (2–3× higher risk than males; lower repolarisation reserve).
- Baseline QTc >450 ms (males) or >470 ms (females).
- Age >65 years.
- Heart failure (LVEF <35%) or left ventricular hypertrophy.
- Bradycardia (resting HR <60 bpm).
- Electrolyte abnormalities (hypokalaemia, hypomagnesaemia, hypocalcaemia).
- Hepatic impairment (reduced drug metabolism).
- Polypharmacy (≥5 medications).
- Family history of LQTS or SCD (may harbour latent congenital LQTS).
Risk Stratification & ECG Monitoring
The Schwartz Score for Congenital LQTS
The modified Schwartz score remains the principal clinical tool for estimating the probability of congenital LQTS in the absence of genetic testing results.
| Criterion | Points |
|---|---|
| ECG findings | |
| QTc ≥480 ms | 3 |
| QTc 460–479 ms (males 450–459 ms) | 2 |
| QTc 440–459 ms | 1 |
| Torsades de pointes (TdP) | 2 |
| T-wave alternans | 1 |
| Notched T wave in 3 leads | 1 |
| Low heart rate for age | 0.5 |
| Clinical history | |
| Syncope with stress (LQTS1/2) | 2 |
| Syncope without stress (LQTS3) | 1 |
| Congenital deafness | 0.5 |
| Family history | |
| Family member with confirmed LQTS | 1 |
| Unexplained SCD in immediate family member <30 years | 0.5 |
Interpretation: ≤1 point = low probability; 1.5–3 points = intermediate probability; ≥3.5 points = high probability. A score ≥3.5 warrants referral to a cardiologist with inherited arrhythmia expertise and genetic testing.
ECG Parameters & Monitoring
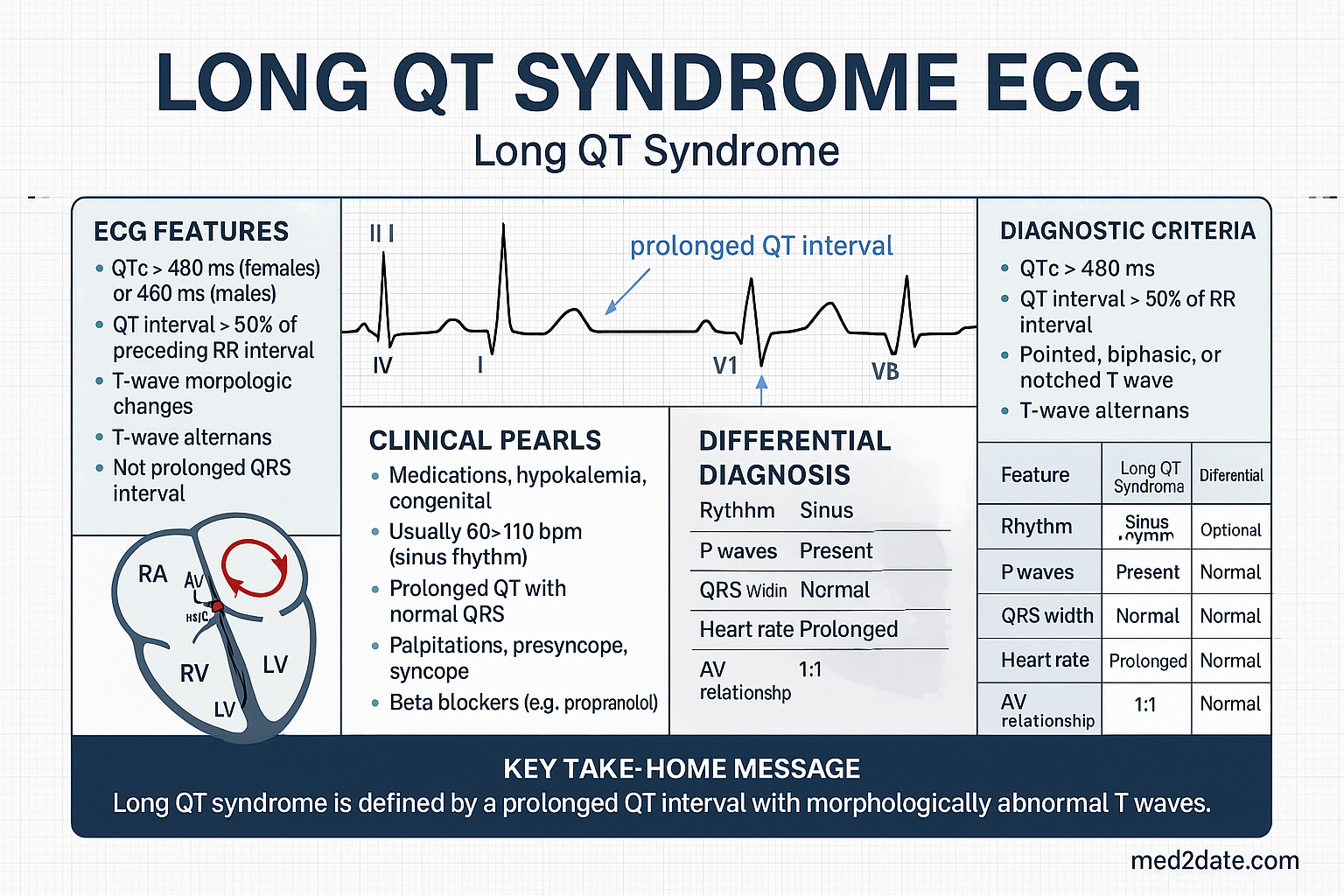
Accurate QTc measurement is the cornerstone of LQTS diagnosis and monitoring:
- QTc formula: Bazett's correction (QTc = QT / √RR) is the standard but overcorrects at heart rates >100 bpm and undercorrects at <60 bpm. Fridericia's formula (QTc = QT / ∛RR) is more accurate at extremes of heart rate and should be used in drug safety studies.
- Measurement technique: Use lead II or V5; measure from the onset of the QRS to the end of the T wave (return to T-P baseline). In bifid T waves, include the second component if the amplitude is ≥50% of the first.
- Normal values: QTc <450 ms (males), <460 ms (females); borderline 450–470 ms (males), 460–480 ms (females); prolonged >470 ms (males), >480 ms (females).
- Exercise stress test: Particularly useful for LQTS1. QTc prolongation during the recovery phase (minutes 1–4 post-exercise) is a hallmark. The maximum QTc during recovery >460 ms has high sensitivity for LQTS1.
- Holter monitoring: 24–48 hour Holter assesses diurnal QTc variation and captures T-wave morphology changes. LQTS3 patients may show nocturnal QTc prolongation.
- Epinephrine (adrenaline) provocation test: IV epinephrine infusion may unmask LQTS1 (paradoxical QTc prolongation) but requires specialist cardiac monitoring facilities and is not universally endorsed in Australia.
Risk Stratification for Cardiac Events
Management: Beta-Blockers, ICD, & Drug Avoidance
Management of LQTS requires a multimodal approach combining pharmacotherapy, device therapy, lifestyle modification, and rigorous avoidance of QT-prolonging agents. Therapy should be individualised based on LQTS genotype, symptom burden, QTc duration, and patient-specific risk factors.
Beta-Blocker Therapy
Beta-blockers are first-line therapy for all congenital LQTS subtypes. They reduce adrenergic-triggered arrhythmias by attenuating sympathetic stimulation of ventricular myocytes and reducing heart rate variability. Non-selective beta-blockers with sustained plasma levels are preferred.
LQTS3-Specific Considerations
LQTS3 (SCN5A gain-of-function) responds less well to beta-blockers, as events are often bradycardia-dependent and occur at rest or during sleep. Sodium channel blockade with mexiletine (not PBS-listed; requires Section 19 importation) or ranolazine (used off-label) may be considered as adjunctive therapy in LQTS3 patients with recurrent events. Flecainide has also been used in LQTS3 with the Brugada overlap phenotype but requires specialist supervision.
Implantable Cardioverter-Defibrillator (ICD)
ICD implantation is a life-saving intervention for high-risk LQTS patients:
- Class I indication: Survivors of cardiac arrest (secondary prevention).
- Class IIa indication: Recurrent syncope on maximised beta-blocker therapy; QTc >500 ms with additional risk factors.
- Class IIb indication: High-risk genotype (e.g., LQTS2 with early onset, LQTS3 with QTc >500 ms) despite compliance with beta-blockers; consideration in patients unable to take beta-blockers.
ICD programming should incorporate long detection intervals to avoid inappropriate shocks for self-terminating TdP. Anti-tachycardia pacing (ATP) as first therapy is preferred. ICD implantation is performed at major Australian cardiac centres; costs are covered under Medicare for approved indications.
Left Cardiac Sympathetic Denervation (LCSD)
LCSD (video-assisted thoracoscopic sympathectomy of the lower half to one-third of the left stellate ganglion and T2–T4 ganglia) reduces sympathetic innervation to the heart. Indications include:
- Recurrent syncope or TdP on maximised beta-blocker therapy.
- Beta-blocker intolerance or contraindication.
- Patients refusing or not meeting criteria for ICD.
- ICD patients with recurrent appropriate shocks (adjunct to beta-blockers).
LCSD is available at select Australian tertiary centres (Royal Melbourne Hospital, Westmead Hospital). It reduces cardiac event rates by approximately 70–90% but is not curative — patients remain on beta-blockers and require ongoing follow-up.
Acute Management of Torsades de Pointes
Pharmacological management of recurrent/persistent TdP:
Drug Avoidance & QT-Drug Lists
All patients with congenital or acquired LQTS must maintain strict avoidance of QT-prolonging medications. Key strategies:
- Register the patient's LQTS diagnosis on My Health Record with a prominent allergy/alert flag.
- Refer to the CredibleMeds QTDrugs database (crediblemeds.org) — the international gold-standard list categorising drugs as Known Risk, Possible Risk, or Conditional Risk for TdP.
- Educate patients to carry a medical alert bracelet and wallet card listing QT-prolonging drug avoidance.
- Ensure all prescribing clinicians, pharmacists, and emergency departments are aware of the diagnosis.
- Common drugs to AVOID: ondansetron, domperidone, droperidol, haloperidol IV, methadone, fluoroquinolones (moxifloxacin, ciprofloxacin), erythromycin IV, azithromycin in high doses, amitriptyline, fluconazole in high doses, chloroquine.
- Safe antiemetics: metoclopramide, prochlorperazine (Stemetil®). Safe antibiotics: amoxicillin, cephalexin (Keflex®), trimethoprim-sulfamethoxazole (use with K⁺ monitoring).
Lifestyle Modifications
- LQTS1: Avoid competitive swimming and diving (high event risk); recreational swimming only with a buddy present. Avoid sudden startling noises (alarm clocks).
- LQTS2: Minimise exposure to startle stimuli; emotional stress management; caution during the post-partum period (elevated risk of cardiac events).
- All subtypes: Maintain adequate hydration and electrolyte intake; avoid excessive alcohol; treat fever aggressively (fever lowers IKr function and prolongs QT); avoid extreme temperatures (very hot baths/saunas).
- Exercise: Most patients on beta-blockers can participate in recreational and competitive sport with appropriate counselling and genotype-specific advice. The 2015 ESC recommendations and subsequent CSANZ guidance allow competitive sport for genotype-positive/phenotype-negative patients and carefully selected phenotype-positive patients on therapy.
- Pregnancy: Continue beta-blockers throughout pregnancy; nadolol and propranolol are preferred. Labetalol may be used. Monitor fetal growth (beta-blockers may cause small-for-gestational-age infants). Post-partum period is high-risk for LQTS2.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Aboriginal and Torres Strait Islander peoples experience higher rates of cardiovascular disease and sudden cardiac death compared with non-Indigenous Australians. Inherited arrhythmia syndromes, including LQTS, may contribute to the elevated burden of sudden cardiac death in young Indigenous Australians, though epidemiological data specific to LQTS in this population remain limited. The AIHW reports that cardiovascular disease is the leading cause of the life expectancy gap, and genetic cardiac conditions likely remain underdiagnosed.
📚 References
- 1. Schwartz PJ, Crotti L, Insolia R. Long QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5(4):868–877.
- 2. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10(12):1932–1963.
- 3. Ackerman MJ, Priori SG, Dubin AM, et al. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: are all beta-blockers equivalent? Heart Rhythm. 2017;14(1):e41–e44.
- 4. Giudicessi JR, Ackerman MJ. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr Probl Cardiol. 2013;38(10):417–455.
- 5. Cardiac Society of Australia and New Zealand. Guidelines for the management of inherited arrhythmia syndromes. Heart Lung Circ. 2017;26(1):1–32.
- 6. Woosley RL, Black K, Heise CW, Romero K. CredibleMeds.org: what does it offer? Trends Cardiovasc Med. 2018;28(2):94–99.
- 7. Australian Institute of Health and Welfare (AIHW). Cardiovascular disease in Aboriginal and Torres Strait Islander people. AIHW Cat. No. CVD 79. Canberra: AIHW; 2023.
- 8. Skinner JR, Crawford J, Smith W, et al. Prospective, population-based long QT molecular autopsy study of post-mortem negative sudden death in 1 to 40 year olds. Heart Rhythm. 2011;8(3):412–419.
- 9. SADS Foundation Australia. Sudden Arrhythmic Death Syndrome: clinical resources and family support. Available at: sads.org.au. Accessed 2024.
- 10. Peltenburg B, Krijnen WP, den Ruijter HM, et al. Sex differences in drug-induced QTc prolongation: a systematic review and meta-analysis. Front Cardiovasc Med. 2022;9:888403.
- 11. Al-Khatib SM, LaPointe NMA, Kramer JM, Califf RM. What clinicians should know about the QT interval. JAMA. 2003;289(16):2120–2127.
- 12. Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350(10):1013–1022.
- 13. Members Service Providers (MBS). Medicare Benefits Schedule — Item 73291 (Genomic sequencing). Australian Government Department of Health and Aged Care. Available at: mbsonline.gov.au. Accessed 2024.
- 14. Royal Australasian College of Physicians (RACP). Genetic evaluation of sudden cardiac death: position statement. Sydney: RACP; 2022.
- 15. Pelliccia A, Sharma S, Gati S, et al. 2020 ESC guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J. 2021;42(1):17–96.