📋 Key Information Summary
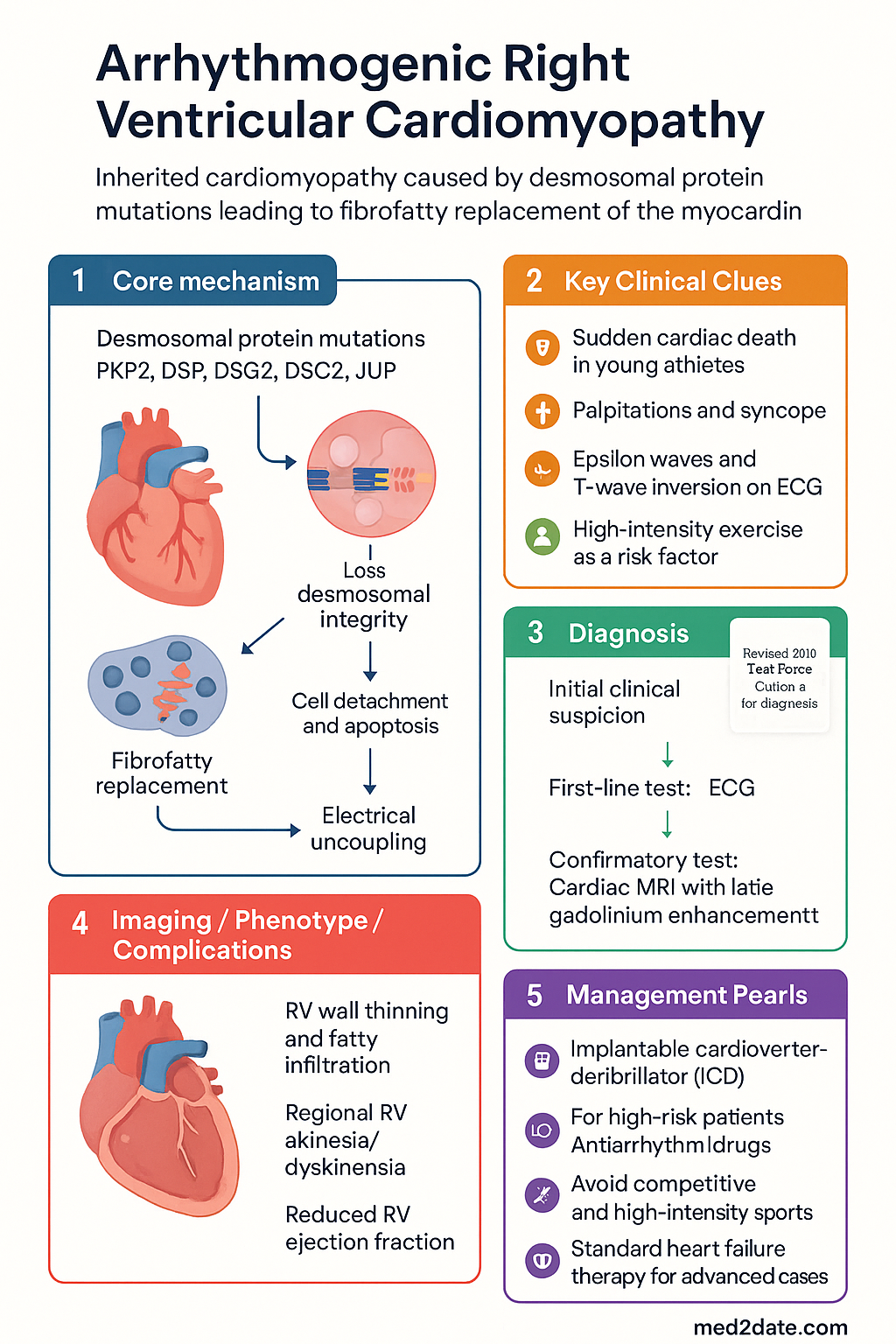
- ARVC is an inherited cardiomyopathy caused by desmosomal protein mutations (PKP2, DSP, DSG2, DSC2, JUP), leading to fibrofatty replacement of the right ventricular myocardium.
- Predominant mode of inheritance is autosomal dominant with variable penetrance and expressivity; compound/digenic heterozygotes have more severe disease.
- ARVC is an important cause of sudden cardiac death (SCD) in young athletes aged <35 years; exercise and high-intensity sport are potent disease modifiers.
- The revised 2010 Task Force Criteria (TFC) classify diagnosis as definite, borderline, or possible based on major and minor criteria across six categories.
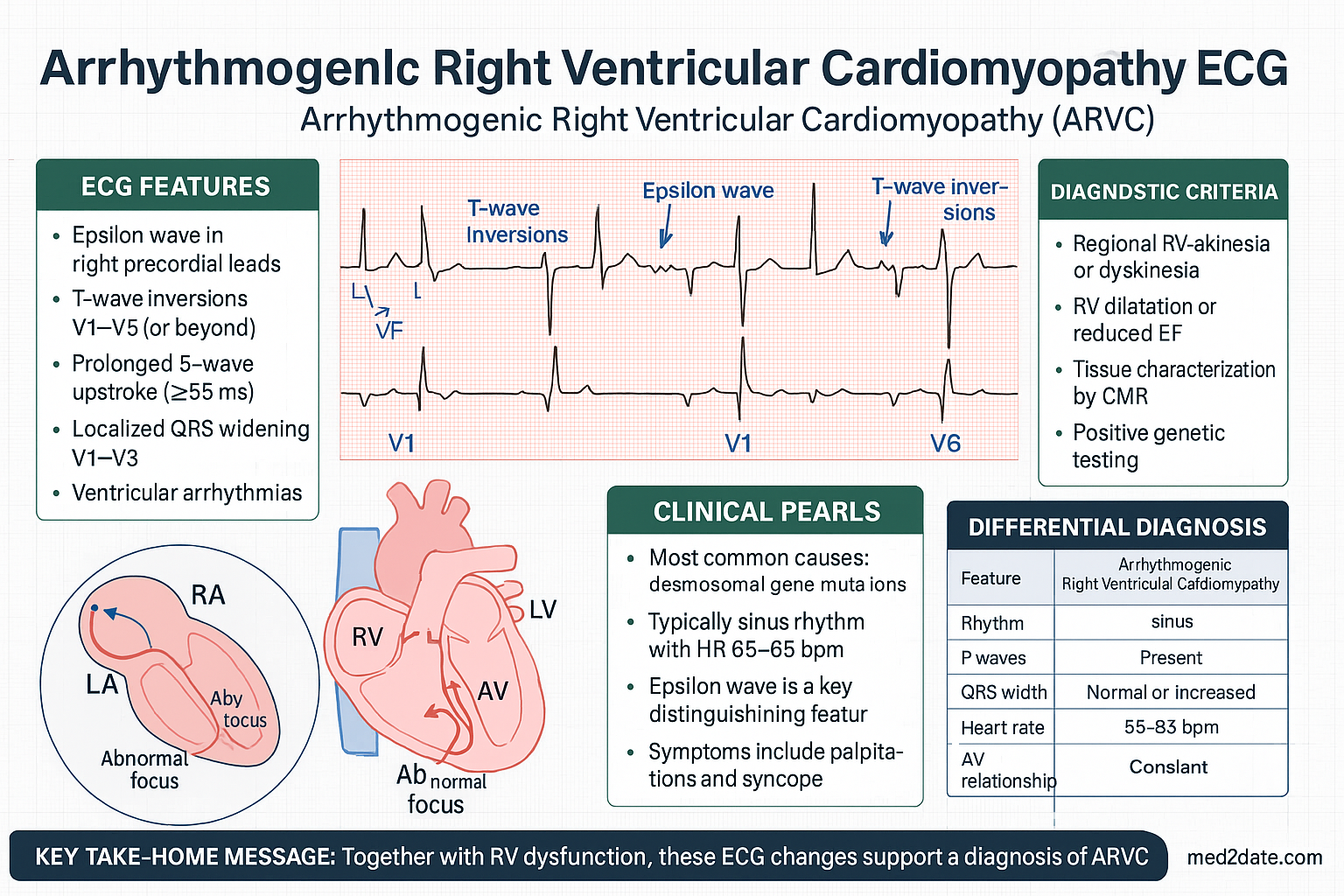
- Classic ECG findings include epsilon waves, T-wave inversion in V1–V3 (age >14), and prolonged S-wave upstroke ≥55 ms in V1–V3.
- Cardiac MRI with late gadolinium enhancement is the gold-standard imaging modality, demonstrating RV wall thinning, fatty infiltration, regional RV akinesia/dyskinesia, and reduced RV ejection fraction.
- All patients should undergo genetic testing targeting a panel of at least the five core desmosomal genes; cascade screening of first-degree relatives is mandatory.
- Implantable cardioverter-defibrillator (ICD) implantation is indicated for survivors of ventricular fibrillation/sustained VT (Class I) and selected high-risk patients (Class IIa).
- Antiarrhythmic drug therapy includes sotalol (first-line) or amiodarone; catheter ablation is reserved for recurrent ICD shocks or drug-refractory VT.
- Complete cessation of competitive and high-intensity endurance sport is strongly recommended regardless of genotype to slow disease progression.
- Management of heart failure in ARVC follows standard HF therapy (ACE inhibitor/ARB, beta-blocker, diuretics); advanced cases may require cardiac transplantation.
- Aboriginal and Torres Strait Islander peoples face barriers including limited access to genetic testing, cardiac MRI, and specialist electrophysiology services in remote areas.
Introduction & Australian Epidemiology
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heritable cardiomyopathy characterised by progressive fibrofatty replacement of the right ventricular myocardium. First described by Fontaine and colleagues in 1977, ARVC is now recognised as one of the leading causes of sudden cardiac death (SCD) in individuals under 35 years of age, particularly among competitive athletes.
The estimated prevalence in the general population ranges from 1 in 2,000 to 1 in 5,000. In Australia, ARVC accounts for an estimated 10–15% of SCD cases in young athletes referred to the Victorian Institute of Forensic Medicine and is consistently identified in national SCD registries. The prevalence appears higher in certain geographic regions, including the Veneto region of Italy (from which early case series originated), and in populations with high rates of consanguinity.
ARVC typically manifests in adolescence or early adulthood (mean age at diagnosis 25–35 years), with a male predominance for symptomatic arrhythmias (male-to-female ratio approximately 3:1). Female patients tend to present later and more often with heart failure than arrhythmia. Disease expression is influenced by genotype (number of affected desmosomal genes, specific mutation type), exercise exposure, and sex.
In the Australian context, the Cardiac Inherited Diseases Registry (previously based at the Baker Heart and Diabetes Institute, now expanding nationally) and the Coroner's Court SCD review process have been instrumental in identifying affected families. Access to specialist cardiomyopathy and genetic counselling services varies significantly between metropolitan and remote Australia.
Genetics & Pathophysiology (Desmosomal Mutations)
Molecular Genetics
ARVC is predominantly caused by pathogenic variants in genes encoding desmosomal proteins. The desmosome is a cell–cell adhesion complex critical for mechanical and electrical coupling of cardiomyocytes.
| Gene | Protein | Frequency of Pathogenic Variants | Key Phenotypic Features |
|---|---|---|---|
| PKP2 | Plakophilin-2 | 30–43% of genotype-positive cases | Most common; classic ARVC phenotype; generally milder than compound/digenic |
| DSP | Desmoplakin | 10–15% | Biventricular or left-dominant disease (ARVC/D-Like); high SCD risk; skin/woolly hair associations |
| DSG2 | Desmoglein-2 | 5–10% | Variable penetrance; may overlap with DCM phenotype |
| DSC2 | Desmocollin-2 | 2–5% | Rare; autosomal recessive forms described |
| JUP | Junction plakoglobin (γ-catenin) | <2% | Naxos disease in autosomal recessive form (palmar-plantar keratoderma, woolly hair) |
Non-Desmosomal Genes
Approximately 40–50% of patients meeting Task Force Criteria have no identifiable desmosomal mutation. Non-desmosomal genes associated with ARVC or ARVC-like phenotypes include TMEM43 (ARVC-5, p.S358L founder mutation in Newfoundland), PLN (phospholamban — hot-spot R14del variant), DES (desmin), TTN, RYR2, and LMNA. Whole-exome and whole-genome sequencing panels are increasingly used in Australian diagnostic laboratories.
Inheritance Patterns
- Autosomal dominant (most common): Variable penetrance (60–70% by age 60); compound heterozygosity or digenic inheritance in ~5–10% of cases confers earlier onset and worse prognosis.
- Autosomal recessive: Naxos disease (JUP), Carvajal syndrome (DSP) — rare; associated with ectodermal features (woolly hair, palmoplantar keratoderma).
- De novo mutations: Account for ~15–20% of cases; no family history despite genetic testing of parents.
Pathophysiology
Loss of desmosomal integrity leads to:
- Cell detachment and apoptosis: Mechanical stress (especially during exercise) causes cardiomyocyte death, particularly in the thin-walled right ventricle where wall stress is highest.
- Fibrofatty replacement: Adipogenesis is triggered by aberrant Wnt/β-catenin signalling; dead myocardium is replaced by fibrous tissue and adipocytes. This process is patchy, affecting the RV "triangle of dysplasia" (inflow tract, apex, outflow tract).
- Electrical uncoupling: Fibrofatty scar creates zones of slow conduction and conduction block, forming the substrate for re-entrant ventricular tachycardia.
- Gap junction remodelling: Desmosomal mutations impair trafficking and localisation of connexin-43 (Cx43) to intercalated discs, further disrupting electrical coupling.
Clinical Features & Task Force Criteria
Clinical Phases
ARVC progresses through four clinical phases, though not all patients progress through every stage:
Presenting Symptoms
- Palpitations: Most common presenting symptom; often triggered by exercise
- Syncope / pre-syncope: Occurs in ~30% at presentation; associated with higher risk of SCD
- SCD: May be the first manifestation in up to 20% of cases
- Heart failure symptoms: Dyspnoea, peripheral oedema — typically late presentations
- Chest pain: Atypical, may relate to myocardial inflammation
Revised 2010 Task Force Criteria
The 2010 International Task Force Criteria are the current diagnostic standard. Diagnosis is based on a points system across six categories: (1) Global or regional dysfunction and structural alterations, (2) Tissue characterisation, (3) Repolarisation abnormalities, (4) Depolarisation/conduction abnormalities, (5) Arrhythmias, and (6) Family history. Points are scored as 1 (major) or 0.5 (minor).
| Category | Major Criteria (1 pt) | Minor Criteria (0.5 pt) |
|---|---|---|
| I. RV dysfunction | Regional RV akinesia, dyskinesia, or aneurysm + mild–severe RV dilation or reduced RVEF | Regional RV akinesia/dyskinesia + mild RV dilation or reduced RVEF; OR absence of (or only mild) RV dilation |
| II. Tissue characterisation | Residual myocytes <60% (or <50% if family history positive) with fibrous replacement on EMB, in ≥1 sample | Residual myocytes 60–75% (or 50–65% if family history positive) with fibrous replacement |
| III. Repolarisation | Right precordial T-wave inversion (V1–V3) in absence of RBBB, age ≥14 | T-wave inversion V1–V2 in absence of RBBB (age ≥14), OR T-wave inversion V1–V4 in presence of complete RBBB |
| IV. Depolarisation | Epsilon wave in V1–V3 | Late potentials on SAECG; OR filtered QRS ≥114 ms; OR duration of terminal QRS <40 μV ≥38 ms; OR root-mean-square voltage of last 40 ms ≤20 μV; OR S-wave upstroke ≥55 ms in V1–V3 |
| V. Arrhythmias | Non-sustained or sustained VT of LBBB morphology with superior axis | Non-sustained or sustained VT of RV outflow configuration, LBBB morphology with inferior axis; OR >500 PVCs/24 h on Holter |
| VI. Family history | ARVC confirmed in first-degree relative (TFC); OR first-degree relative SCD <35 years with suspected ARVC; OR pathogenic mutation identification | Family history of ARVC in more distant relative; OR family history of SCD <35 years in second-degree relative |
Diagnostic thresholds:
- Definite ARVC: 2 major, or 1 major + 2 minor, or 4 minor criteria across different categories
- Borderline ARVC: 1 major + 1 minor, or 3 minor criteria
- Possible ARVC: 1 major, or 2 minor criteria
Investigations (ECG, MRI, Genetics)
Resting 12-Lead ECG
The ECG is abnormal in up to 90% of patients with overt ARVC and forms the basis of several Task Force criteria.
Echocardiography
Transthoracic echocardiography is the first-line imaging modality. Findings may include regional RV wall motion abnormalities (aneurysm, dyskinesia), RVOT dilation (>32 mm parasternal or >36 mm PLAX), and reduced TAPSE (<17 mm). However, echocardiography has limited sensitivity for early/mild disease compared to cardiac MRI.
Cardiac MRI
Key MRI findings in ARVC:
- Regional RV wall motion abnormalities (akinesia, dyskinesia, aneurysm) — typically affecting the sub-tricuspid area, RV apex, and RVOT (triangle of dysplasia)
- RV dilatation and reduced RV ejection fraction (<40%)
- Intra-myocardial fatty infiltration (T1-weighted spin-echo sequences with fat suppression)
- LGE of the RV wall or LV myocardium indicating fibrosis
- LV involvement: LGE in the inter-ventricular septum or LV free wall in up to 75% of patients
Australian availability: Cardiac MRI is available at all tertiary centres and many private radiology practices. MBS Item 63448 (MRI heart, structural) requires specific indication; prior echocardiography and/or ECG findings typically support the request. MRI with LGE requires a specialist radiologist reporting.
Genetic Testing
Endomyocardial Biopsy
Right ventricular endomyocardial biopsy (EMB) provides histological confirmation (fibrofatty replacement, <60% residual myocytes) and is a major TFC criterion when positive. However, EMB has limited sensitivity due to the patchy distribution of disease and the risk of RV perforation. It is reserved for diagnostic uncertainty when non-invasive investigations are inconclusive, and should be performed at experienced centres only.
Investigations Summary
| Investigation | Sensitivity for ARVC | Key Role | MBS Item |
|---|---|---|---|
| Resting ECG | ~90% (overt disease) | Initial screening, TFC criteria | 11707 |
| Echocardiography | ~60–75% | First-line imaging, serial follow-up | 55118 |
| Cardiac MRI | ~90–95% | Gold standard; tissue characterisation | 63448 |
| Holter Monitor | ~80% (PVC detection) | Arrhythmia burden, VT morphology | 11709 |
| SAECG | ~50–80% | Late potentials, depolarisation criteria | 11710 |
| Genetic Testing | ~50–60% | Diagnosis confirmation, cascade screening | 73296 |
| EMB | ~65–70% | Histological confirmation (selective use) | 38235 |
Management (ICD, Antiarrhythmics, Activity Restriction)
General Principles
Management of ARVC centres on three pillars: (1) prevention of sudden cardiac death (ICD, activity restriction), (2) suppression of ventricular arrhythmias (antiarrhythmic drugs, catheter ablation), and (3) management of progressive heart failure (standard HF therapy, transplant assessment).
Implantable Cardioverter-Defibrillator (ICD)
| Indication | Class | Details |
|---|---|---|
| Survivor of cardiac arrest (VF/sustained VT) | I | Strongest indication; secondary prevention |
| Syncope presumed due to VT/VF | IIa | When other causes excluded; consider electrophysiology study |
| Non-sustained VT + additional risk factors | IIa | Risk factors: male, compound/digenic mutations, significant RV dysfunction, LV involvement |
| Asymptomatic genotype-positive with risk factors | IIb | Compound/digenic genotype, competitive athlete, familial SCD history |
| Asymptomatic, single mutation, no structural disease | III (not indicated) | Annual surveillance rather than ICD |
Device considerations: Subcutaneous ICD (S-ICD) is increasingly used to avoid lead-related complications in the dilated and dysfunctional RV. Transvenous ICD with right-sided implantation remains an option. Anti-tachycardia pacing (ATP) programming should be cautious as aggressive pacing may accelerate ARVC. Programming prolonged detection intervals reduces inappropriate shocks. Remote monitoring is standard in Australia.
Antiarrhythmic Drug Therapy
Catheter Ablation
Catheter ablation is indicated for recurrent VT causing ICD shocks or drug-refractory VT. Epicardial ablation is often necessary as the substrate in ARVC frequently involves the sub-epicardial layer. Success rates are 60–70% at experienced centres, but recurrence rates remain high (~30–50%) due to progressive fibrofatty replacement. Endocardial–epicardial combined approaches achieve better outcomes. Referral to a high-volume electrophysiology centre is recommended.
Activity Restriction
Heart Failure Management
Progressive RV and biventricular failure is managed according to standard heart failure guidelines:
- ACE inhibitor (e.g., ramipril 2.5–10 mg daily) or ARB (e.g., valsartan 40–320 mg BD)
- Beta-blocker (carvedilol 3.125–25 mg BD or bisoprolol 1.25–10 mg daily) — also for arrhythmia suppression
- Diuretics for symptomatic fluid overload (frusemide 20–80 mg daily, spironolactone 25–50 mg daily)
- Cardiac resynchronisation therapy (CRT) may be considered for biventricular failure with wide QRS >150 ms
- Cardiac transplantation referral for refractory heart failure (conduit to transplant established early)
Treatment Algorithm
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Aboriginal and Torres Strait Islander peoples may experience delayed diagnosis and suboptimal management of inherited cardiac conditions including ARVC, due to systemic barriers in access to specialist cardiac and genetic services. While ARVC prevalence data specific to Indigenous Australians are limited, cardiovascular disease burden is significantly higher in this population, and inherited cardiomyopathies should not be overlooked.
📚 References
- 1. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Circulation. 2010;121(13):1533-1541.
- 2. Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61-72.
- 3. Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Circulation. 2018;138(13):e272-e391.
- 4. Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16(11):e301-e372.
- 5. James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62(14):1290-1297.
- 6. Groeneweg JA, Bhonsale A, James CA, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015;8(3):437-446.
- 7. Rizzo S, Pilichou K, Thiene G, Basso C. The changing spectrum of arrhythmogenic (right ventricular) cardiomyopathy. Cell Tissue Res. 2015;360(3):481-496.
- 8. Miles C, Finocchiaro G, Papadakis M, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation. 2019;139(15):1786-1797.
- 9. Wijnmaalen AP, Zeppenfeld K, de Riva M, et al. Endocardial/epicardial versus epicardial-only catheter ablation for ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy. Europace. 2022;24(7):1150-1159.
- 10. Australian Institute of Health and Welfare (AIHW). Cardiovascular disease in Aboriginal and Torres Strait Islander people. Canberra: AIHW; 2023. Available at: aihw.gov.au.
- 11. Macfarlane PW, Antzelevitch C, Haissaguerre M, et al. The early repolarization pattern: a consensus paper. J Am Coll Cardiol. 2015;66(4):470-477.
- 12. Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice (Red Book). 10th edn. East Melbourne: RACGP; 2018.