📋 Key Information Summary
- Restrictive cardiomyopathies (RCM) are characterised by abnormal diastolic function with normal or reduced ventricular volumes; infiltrative cardiomyopathies involve extracellular or intracellular substance deposition within the myocardium.
- Cardiac amyloidosis — AL (light-chain) and ATTR (transthyretin) — is the most common infiltrative cause; Tc-99m pyrophosphate scintigraphy distinguishes ATTR from AL when serum free light chains and urine immunofixation are negative.
- ATTR cardiac amyloidosis (both wild-type and hereditary) is increasingly recognised in Australia, particularly in males >65 years with heart failure and prior carpal tunnel syndrome.
- Tafamidis (Vyndaqel®) is PBS-listed (Authority Required) for ATTR cardiomyopathy and reduces all-cause mortality and cardiovascular hospitalisations.
- AL amyloidosis requires urgent haematology referral; first-line treatment is bortezomib-based chemotherapy (CyBorD) with consideration of autologous stem cell transplant.
- Cardiac sarcoidosis presents with heart failure, conduction disease, or ventricular arrhythmias; FDG-PET/CT is the key diagnostic and monitoring tool, and immunosuppression with corticosteroids is first-line.
- Hereditary haemochromatosis (HFE-related) is common in Australians of Northern European descent; cardiac iron overload causes dilated or restrictive cardiomyopathy and is detected by cardiac MRI T2* mapping.
- Iron chelation with deferasirox or deferoxamine, combined with serial venesection, reverses cardiac iron loading if initiated before irreversible fibrosis.
- Fabry disease (α-galactosidase A deficiency) causes LVH mimicking HCM with restrictive physiology; enzyme replacement therapy (agalsidase beta) and chaperone therapy (migalastat) are available.
- Endomyocardial fibrosis is rare in Australia but seen in some migrant communities from tropical regions; glycogen storage disorders (PRKAG2, Danon disease) cause severe paediatric cardiomyopathy.
- All patients with infiltrative cardiomyopathy require genetic counselling and cascade screening where an hereditary aetiology is confirmed.
- Aboriginal and Torres Strait Islander peoples have higher rates of iron overload syndromes and rheumatic heart disease, which may mimic or coexist with infiltrative cardiomyopathy; culturally safe cardiac screening is essential.
Introduction & Australian Epidemiology
Restrictive cardiomyopathies (RCM) represent a heterogeneous group of myocardial diseases characterised by increased ventricular stiffness, impaired diastolic filling with preserved or reduced ventricular volumes, and ultimately elevated filling pressures. Infiltrative cardiomyopathies constitute a major subset in which specific substances — either extracellular (amyloid, fibrosis) or intracellular (iron, glycosphingolipids, glycogen) — accumulate within the myocardium, disrupting normal architecture and function.
In Australia, the epidemiology of infiltrative cardiomyopathies has shifted significantly over the past decade, driven by improved diagnostic imaging and heightened clinical awareness:
- Cardiac amyloidosis: Estimated prevalence of wild-type ATTR (ATTRwt) is 16–25% in autopsy studies of elderly males with heart failure. The Australian Amyloidosis Network reports increasing referral rates since the availability of Tc-99m PYP imaging and disease-modifying therapies.
- Hereditary ATTR: The Val122Ile variant is found in ~3–4% of African-descent populations; other TTR variants (Val30Met, Thr60Ala) occur in Australian families of Celtic and other European ancestry. Genetic testing through national services (e.g., Victorian Clinical Genetics Services) is available.
- Cardiac sarcoidosis: Sarcoidosis prevalence in Australia is estimated at 15–20 per 100,000; cardiac involvement occurs in 5–10% of systemic sarcoidosis cases, though autopsy studies suggest subclinical cardiac involvement in up to 25%.
- Hereditary haemochromatosis: The C282Y homozygous HFE mutation affects approximately 1 in 200 Australians of Northern European descent, making it one of the most common autosomal recessive conditions in Australia. Cardiac iron overload occurs in 15–20% of untreated homozygotes, typically presenting between ages 30–50.
- Fabry disease: Estimated incidence 1 in 40,000–117,000 live births; cardiac involvement is the leading cause of death. Newborn screening pilot programmes have been conducted in some Australian states.
- Endomyocardial fibrosis: Rare in the general Australian population but seen in migrant communities from sub-Saharan Africa, South Asia, and Latin America.
The Australian Institute of Health and Welfare (AIHW) data show that cardiomyopathies contribute to approximately 5,500 hospitalisations per year nationally. Early identification and accurate subtyping of infiltrative cardiomyopathies is critical because specific disease-modifying therapies now exist for many conditions, and treatment outcomes are markedly better when initiated before irreversible myocardial damage occurs.
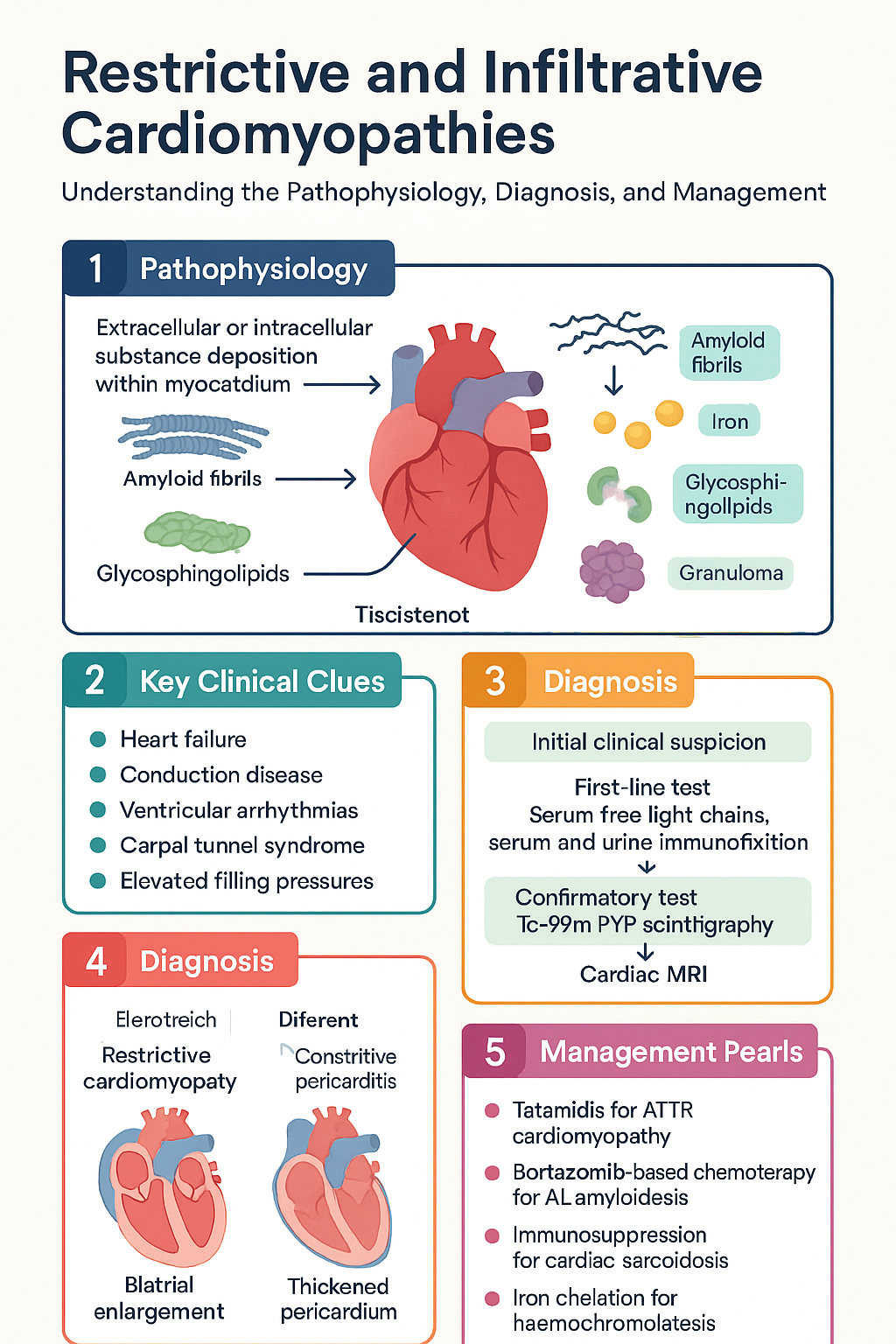
Pathophysiology of Restrictive & Infiltrative Cardiomyopathies
The restrictive phenotype results from increased myocardial stiffness that impairs ventricular filling during diastole. This may be caused by:
- Extracellular infiltration: Amyloid fibrils (AL or ATTR) deposit in the interstitium, displacing cardiomyocytes and causing wall thickening with a stiff, non-compliant ventricle. Endomyocardial fibrosis (EMF) involves progressive fibrotic obliteration of the ventricular apex.
- Intracellular infiltration: Iron (haemochromatosis) deposits within cardiomyocyte lysosomes and mitochondria, generating reactive oxygen species and causing oxidative damage. Glycosphingolipids (Fabry disease) accumulate in lysosomes due to deficient α-galactosidase A activity. Glycogen (PRKAG2, Danon disease) accumulates within lysosomes and cytoplasm.
- Granulomatous infiltration: Non-caseating granulomas in cardiac sarcoidosis replace normal myocardium and cause fibrosis, conduction system disruption, and ventricular arrhythmias.
Regardless of aetiology, the haemodynamic consequences converge: elevated ventricular end-diastolic pressures transmit retrogradely to the atria and venous circulation, causing pulmonary congestion and systemic venous congestion. Atrial dilatation predisposes to atrial fibrillation. The "dip-and-plateau" or "square root sign" on cardiac catheterisation is a hallmark of restrictive physiology.
Cardiac Amyloidosis
AL vs ATTR Differentiation
Cardiac amyloidosis is caused by the deposition of misfolded protein fibrils in the myocardium. Accurate typing is mandatory because treatments differ fundamentally between subtypes:
| Feature | AL Amyloidosis | ATTR Amyloidosis (Wild-Type) | ATTR Amyloidosis (Hereditary) |
|---|---|---|---|
| Precursor protein | Immunoglobulin light chains (κ or λ) | Transthyretin (normal sequence) | Mutant transthyretin |
| Age at onset | 50–70 years (median ~60) | >65 years (predominantly male) | 30–60 years (variant-dependent) |
| Sex predominance | M:F ≈ 1:1 | M:F ≈ 15–50:1 | Variable |
| Associated features | Nephrotic syndrome, macroglossia, periorbital purpura, peripheral neuropathy, hepatomegaly | Bilateral carpal tunnel syndrome, spinal stenosis, HFpEF in elderly male | Peripheral/autonomic neuropathy, cardiac involvement (variant-dependent) |
| Serum free light chains | Abnormal κ/λ ratio | Normal | Normal |
| Serum/urine immunofixation | Monoclonal protein detected | Negative | Negative |
| Tc-99m PYP scintigraphy | Negative (or weakly positive with confirmed non-AL) | Strongly positive (Perugini grade 2–3) | Strongly positive |
| Tissue biopsy confirmation | Positive Congo red + mass spectrometry typing | Positive Congo red + TTR typing; genetic testing | Positive Congo red + TTR gene sequencing |
Technetium-99m Pyrophosphate (Tc-99m PYP) Imaging
Tc-99m PYP (or DPD/HMDP) scintigraphy has transformed the non-invasive diagnosis of ATTR cardiac amyloidosis. The Perugini visual scoring system grades cardiac uptake:
Quantitative analysis using the heart-to-contralateral-lung (H/CL) ratio at 1 hour (≥1.5 on planar imaging or ≥1.3 with SPECT/CT) further improves diagnostic accuracy. Tc-99m PYP imaging is available at most major nuclear medicine centres in Australia, including Royal Melbourne Hospital, Royal Prince Alfred Hospital, Flinders Medical Centre, and Royal Brisbane and Women's Hospital. MBS item 61318 applies for myocardial perfusion studies and related cardiac nuclear medicine.
Cardiac MRI in Amyloidosis
Cardiovascular MRI (CMR) demonstrates characteristic findings:
- Global subendocardial or transmural late gadolinium enhancement (LGE) in a non-coronary distribution
- Elevated native T1 mapping values (>1,050 ms at 1.5T) and extracellular volume (ECV >40%)
- Difficulty nulling the myocardium on inversion-recovery sequences (a "red flag" sign)
- Biventricular wall thickening with small chamber sizes, biatrial enlargement
Treatment: ATTR Cardiac Amyloidosis
Treatment: AL Cardiac Amyloidosis
AL amyloidosis is a haematological emergency with a median survival of <6 months when cardiac involvement is advanced (Mayo Stage IIIb). Treatment is directed at the underlying clonal plasma cell disorder.
Heart Failure Management in Amyloidosis
- Diuretics: Loop diuretics (furosemide 20–80 mg PO daily, torsemide 10–20 mg PO daily) are the mainstay of volume management. Titrate to maintain euvolaemia.
- Anticoagulation: Consider for atrial fibrillation (common) — CHA₂DS₂-VASc scoring applies; direct oral anticoagulants (DOACs) are generally preferred over warfarin.
- Pacemaker/ICD: Conduction disease and ventricular arrhythmias are common in ATTR. Pacemaker implantation for advanced AV block. ICD consideration for secondary prevention of sudden cardiac death; primary prevention role is uncertain.
- Advanced therapies: Heart transplantation may be considered in selected AL patients who achieve haematological complete response, or in hereditary ATTR with combined heart-liver transplant (liver being the source of TTR production).
Cardiac Sarcoidosis
Diagnosis of Cardiac Involvement
Cardiac sarcoidosis (CS) is notoriously difficult to diagnose because endomyocardial biopsy has low sensitivity (~20–30%) due to the patchy nature of granulomatous infiltration. The 2017 Japanese Circulation Society (JCS) criteria and the Heart Rhythm Society (HRS) expert consensus provide the most widely used diagnostic frameworks:
Diagnosis is established by either:
- Histological diagnosis: Non-caseating granulomas on endomyocardial biopsy with no alternative cause identified.
- Clinical diagnosis (JCS criteria): Two or more major criteria, OR one major plus two or more minor criteria from the following:
| Category | Criteria |
|---|---|
| Major | Advanced AV block (including complete heart block) or sustained ventricular tachycardia; basal thinning of the interventricular septum or regional wall motion abnormality on imaging; LGE on CMR in a non-coronary distribution; ⁶⁷Ga or ¹⁸F-FDG uptake in the myocardium |
| Minor | Non-sustained ventricular tachycardia; abnormal ECG (fragmented QRS, ST-T changes, pathological Q waves, or axis deviation); reduced LVEF (<50%); LV diastolic dysfunction; elevated cardiac biomarkers (troponin, BNP/NT-proBNP) |
PET Imaging in Cardiac Sarcoidosis
¹⁸F-FDG PET/CT is the cornerstone of non-invasive diagnosis and disease activity monitoring in CS:
- Preparation: Prolonged fasting (>12 hours) with a high-fat, low-carbohydrate diet for 24–48 hours prior to suppress physiological myocardial glucose uptake. Some centres use unfractionated heparin (50 IU/kg IV) 15 minutes before tracer injection.
- Positive scan: Focal or focal-on-diffuse FDG uptake in the myocardium (SUVmax >2× blood pool) in a non-coronary pattern, typically with perfusion defects on ⁸²Rb or ¹³N-ammonia rest perfusion (indicating scar).
- Sensitivity/Specificity: Sensitivity ~85–89% and specificity ~78–95% for CS when combined with perfusion imaging.
- Serial monitoring: FDG-PET is used to assess treatment response during immunosuppression; reduction in FDG uptake indicates disease control.
Cardiac MRI complements PET: LGE in a mid-myocardial or epicardial distribution (particularly basal inferolateral and inferoseptal walls) is the most sensitive technique for detecting myocardial fibrosis. T2-weighted imaging and T2 mapping identify active oedema/inflammation. CMR is preferred when FDG-PET is unavailable or the scan is equivocal.
Immunosuppression
Arrhythmia Management
Arrhythmias are the leading cause of sudden cardiac death in CS and require aggressive management:
- Conduction disease: High-degree AV block is the most common presentation. Permanent pacemaker implantation is indicated for persistent complete heart block. Conduction may recover with immunosuppression, but permanent pacing is generally recommended given the unpredictable course.
- Ventricular tachycardia: Antiarrhythmic drugs — sotalol (80–160 mg PO BD) or amiodarone (loading 200 mg TDS × 10 days, then 200 mg daily) — are first-line. Catheter ablation is increasingly used for recurrent VT refractory to medical therapy; however, recurrence rates are higher than in ischaemic VT due to the diffuse and progressive nature of granulomatous infiltration.
- ICD implantation: Recommended for patients with sustained VT, cardiac arrest, or LVEF <35%. Consider for LVEF 36–50% with positive electrophysiology study or extensive LGE on CMR (a major predictor of arrhythmic events).
- Atrial fibrillation: Common; rate control with cautious beta-blockade (avoiding excessive negative inotropy), or rhythm control with amiodarone. Anticoagulation per CHA₂DS₂-VASc score.
Haemochromatosis — Cardiac Iron Overload
Diagnosis
Hereditary haemochromatosis (HH) is caused by mutations in the HFE gene (most commonly C282Y homozygosity) leading to inappropriately increased intestinal iron absorption. Cardiac involvement occurs when total body iron exceeds storage capacity:
Iron Chelation Therapy
Cardiac Monitoring Protocols
| Investigation | Frequency | Purpose |
|---|---|---|
| Cardiac MRI with T2* | Every 6–12 months during chelation; annually once T2* >20 ms | Quantify myocardial iron; guide chelation intensity |
| Echocardiography | Baseline; then annually or with symptoms | LVEF, diastolic function, wall thickness, RV function |
| 12-lead ECG | Baseline; every 6 months during active chelation | Low voltage complexes, T-wave changes, arrhythmia detection |
| 24-hour Holter | Annually; more frequently if symptomatic | Atrial fibrillation, non-sustained VT, bradyarrhythmias |
| NT-proBNP / BNP | Every 3–6 months during active treatment | Monitor for heart failure progression |
| Serum ferritin | Monthly during chelation | Target <50 μg/L (or 300 μg/L if combined with phlebotomy) |
| Transferrin saturation | Every 3 months | Confirm adequate iron reduction |
Venesection (Phlebotomy)
Serial venesection remains the primary treatment for HFE-related hereditary haemochromatosis without significant cardiac involvement:
- Induction phase: Remove 500 mL blood (≈200–250 mg iron) weekly until serum ferritin <50 μg/L.
- Maintenance phase: Venesection every 2–4 months to keep ferritin <50 μg/L. Frequency guided by haemoglobin and ferritin monitoring.
- Cardiac benefit: Venesection alone may improve early cardiac iron loading but is insufficient for severe myocardial iron deposition (T2* <10 ms), where chelation is required.
- Availability: Venesection is available through the Australian Red Cross Lifeblood service and most GP practices. Bulk-billed under Medicare.
Other Infiltrative Diseases
Fabry Disease
Fabry disease is an X-linked lysosomal storage disorder caused by deficiency of α-galactosidase A (α-Gal A), leading to accumulation of globotriaosylceramide (Gb3) in cardiomyocytes, endothelial cells, and conduction tissue. Cardiac involvement is the leading cause of death in both classical and later-onset phenotypes.
Cardiac manifestations:
- Concentric left ventricular hypertrophy (LVH) — often misdiagnosed as hypertrophic cardiomyopathy (HCM)
- Restrictive diastolic dysfunction with preserved EF until late stages
- Conduction abnormalities (short PR interval, AV block)
- Atrial fibrillation, ventricular tachycardia, and sudden cardiac death
- Myocardial fibrosis on CMR (LGE in basal inferolateral wall is characteristic)
- Valvular disease (mitral and aortic regurgitation)
Diagnosis: Low α-Gal A enzyme activity in males (leucocyte or plasma assay); genetic testing of GLA gene in both sexes (females may have normal enzyme activity due to X-inactivation). Newborn screening pilot studies in Taiwan and some European countries have identified high prevalence of later-onset variants. In Australia, diagnostic testing is available through Victorian Clinical Genetics Services (VCGS) and SA Pathology.
Glycogen Storage Disorders with Cardiac Involvement
Several glycogen storage disorders cause severe cardiomyopathy in childhood:
| Disorder | Gene | Cardiac Features | Key Extra-Cardiac Features |
|---|---|---|---|
| Pompe disease (GSD II) | GAA | Massive LVH, restrictive physiology, conduction abnormalities; infantile form presents with severe hypertrophic cardiomyopathy and heart failure by 2–6 months | Hypotonia, macroglossia, hepatomegaly, respiratory failure; elevated CK, AST, LDH |
| PRKAG2 syndrome | PRKAG2 | Massive LVH, Wolff-Parkinson-White syndrome, progressive conduction disease requiring pacemaker; glycogen-laden cardiomyocytes | Type 2 diabetes in young adults; relatively preserved systolic function early |
| Danon disease | LAMP2 | Severe hypertrophic cardiomyopathy progressing to dilated cardiomyopathy; ventricular pre-excitation; ventricular arrhythmias; male patients typically require transplantation by age 19 | Skeletal myopathy (elevated CK), intellectual disability (variable), retinal degeneration |
| Forbes disease (GSD III) | AGL | Cardiomyopathy in ~15% (usually hypertrophic); generally milder cardiac course | Hepatomegaly, hypoglycaemia, elevated transaminases; may improve with age |
Endomyocardial Fibrosis (EMF)
Endomyocardial fibrosis is a neglected tropical cardiomyopathy endemic to tropical and subtropical regions (sub-Saharan Africa, South Asia, South America). It is characterised by progressive fibrotic obliteration of the right and/or left ventricular apices with associated AV valve dysfunction.
Epidemiology in Australia: Rare in the general population but should be considered in migrants from endemic regions presenting with heart failure, restrictive physiology, and eosinophilia. No specific Australian prevalence data exist.
Diagnosis:
- Echocardiography: apical obliteration, AV valve regurgitation, thrombus in obliterated apex
- Cardiac MRI: endocardial fibrosis with characteristic apical thrombus; LGE in the subendocardial layer
- Cardiac catheterisation: restrictive physiology with elevated filling pressures
- Endomyocardial biopsy: fibrotic endocardium with eosinophilic infiltration (early stages)
Management:
- Medical: diuretics for congestion; anticoagulation if thrombus present; corticosteroids for active eosinophilic myocarditis (early disease)
- Surgical: endocardectomy with AV valve repair/replacement is the definitive treatment (Davies procedure); requires cardiothoracic referral; outcomes best in experienced centres
- No disease-modifying medical therapy exists; heart transplantation may be considered for end-stage biventricular failure
Clinical Presentation & Diagnostic Criteria
Common Clinical Features of Restrictive Cardiomyopathy
- Exertional dyspnoea (most common presenting symptom)
- Orthopnoea and paroxysmal nocturnal dyspnoea
- Fatigue and exercise intolerance
- Peripheral oedema, ascites (right heart failure)
- Palpitations (atrial fibrillation, ventricular arrhythmias)
- Syncope or pre-syncope
- Symptoms related to underlying systemic disease (e.g., peripheral neuropathy in amyloidosis, skin rash in sarcoidosis, arthralgia in haemochromatosis)
- Elevated JVP with rapid x and y descents
- Kussmaul's sign (paradoxical rise in JVP with inspiration)
- S3 and/or S4 gallop
- Mitral and tricuspid regurgitation murmurs
- Hepatomegaly with hepatojugular reflux
- Peripheral oedema, ascites, pleural effusions
- Low voltage ECG (despite apparent wall thickening on echo — a red flag for amyloidosis)
Key Diagnostic "Red Flags" for Specific Aetiologies
| Clinical Clue | Consider | First-Line Investigation |
|---|---|---|
| Elderly male with HFpEF, bilateral carpal tunnel, low voltage ECG, wall thickening | Wild-type ATTR amyloidosis | SFLC, serum/urine IFE → Tc-99m PYP |
| Heart failure + nephrotic syndrome + macroglossia | AL amyloidosis | SFLC, serum/ urine IFE → bone marrow biopsy |
| Young patient with heart block + bilateral hilar lymphadenopathy | Cardiac sarcoidosis | CMR → FDG-PET/CT |
| Dilated/restrictive cardiomyopathy + bronze skin + diabetes + liver disease | Haemochromatosis | Ferritin, TSAT, HFE gene → cardiac MRI T2* |
| Young male with LVH + acroparaesthesia + angiokeratomas + proteinuria | Fabry disease | α-Gal A enzyme activity, GLA gene sequencing |
| Infant with massive LVH + hypotonia + macroglossia | Pompe disease | GAA enzyme activity, GAA gene testing |
| Migrant from tropical region with heart failure + eosinophilia + apical obliteration | Endomyocardial fibrosis | Echo with apical views → CMR |

Investigations
Risk Stratification & Severity Scoring
AL Amyloidosis — Mayo Clinic Staging (2012 Revised)
Cardiac Sarcoidosis — Risk of Arrhythmic Events
| Risk Factor | Implication |
|---|---|
| LVEF <35% | Indication for ICD (as per standard HF guidelines) |
| LVEF 36–54% | Consider ICD if extensive LGE on CMR or inducible VT on EP study |
| Extensive LGE on CMR (≥2 segments) | Independent predictor of arrhythmic events; warrants close monitoring and lower ICD threshold |
| History of syncope | High risk; evaluate for arrhythmogenic cause; lower threshold for ICD |
| Active inflammation on FDG-PET | Higher arrhythmic risk; intensify immunosuppression; closer monitoring |
Haemochromatosis — Cardiac Risk
| Cardiac T2* | Iron Loading | Clinical Risk | Management Intensity |
|---|---|---|---|
| >20 ms | Normal | Low | Routine monitoring; venesection for systemic iron |
| 10–20 ms | Mild-to-moderate | Moderate | Oral chelation + venesection; repeat T2* in 6 months |
| <10 ms | Severe | High (heart failure, arrhythmia, sudden death) | Intensive IV deferoxamine + oral deferiprone; cardiology monitoring |
Pharmacological Management — General Principles
The management of restrictive and infiltrative cardiomyopathies has two parallel goals: (1) treating the underlying infiltrative disease process, and (2) managing the resultant heart failure and arrhythmias. Disease-specific therapies are detailed in the individual subtopic sections above.
Heart Failure Management — Common Principles
Quick Reference — Disease-Specific Therapies
Monitoring
Special Populations
Pregnancy
Paediatrics
Elderly
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007-1016.
- 2. Gillmore JD, Gane E, Taubel J, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11-21.
- 3. Palladini G, Kastritis E, Maurer MS, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71-80.
- 4. Birnie DH, Sauer WH, Bogun F, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. 2014;11(7):1305-1323.
- 5. Slart RHJA, Glaudemans AWJM, Lancellotti P, et al. A joint procedural position statement on imaging in cardiac sarcoidosis. Eur Heart J Cardiovasc Imaging. 2017;18(10):1073-1089.
- 6. Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22(23):2171-2179.
- 7. Carpenter JP, He T, Kirk P, et al. On T2* magnetic resonance and cardiac iron. Circulation. 2011;123(14):1519-1528.
- 8. Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338-346.
- 9. Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid α-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99-109.
- 10. Mocumbi AO, Yacoub S, Yacoub MH. Neglected tropical cardiomyopathies: II. Endomyocardial fibrosis. Heart. 2008;94(3):384-390.
- 11. Rapezzi C, Merlini G, Giansante C, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2009;14(5):281-298.
- 12. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404-2412.
- 13. Kasper EK, Agema WR, Hutchins GM, et al. The causes of dilated cardiomyopathy: a clinicopathologic review of 673 consecutive patients. J Am Coll Cardiol. 1994;23(3):586-590.
- 14. Australian Institute of Health and Welfare (AIHW). Cardiovascular disease in Aboriginal and Torres Strait Islander peoples. AIHW Cat. No. CVD 78. Canberra: AIHW; 2023.
- 15. National Heart Foundation of Australia and Cardiac Society of Australia and New Zealand. Guidelines for the prevention, detection, and management of heart failure in Australia. Med J Aust. 2018;208(10):435-442.
- 16. RHDAustralia (RHD Australia). The 2020 Australian guideline for prevention, diagnosis and management of acute rheumatic fever and rheumatic heart disease. 3rd ed. Darwin: Menzies School of Health Research; 2020.
- 17. Pieroni M, Chimenti C, De Cobelli F, et al. Fluorine-18-fluorodeoxyglucose positron emission tomography identifies the active disease in cardiac sarcoidosis. J Am Coll Cardiol. 2006;47(7):1485-1487.
- 18. Bulut A, Akyıldız ZI, Arslan B, et al. Diagnostic and prognostic value of CMR in cardiac amyloidosis: a meta-analysis. JACC Cardiovasc Imaging. 2023;16(8):1110-1122.
- 19. McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. 2017;121(7):722-730.
- 20. Wechalekar AD, Gillmore JD, Bird J, et al. Guidelines on the management of AL amyloidosis. Br J Haematol. 2015;168(2):186-206.