📋 Key Information Summary
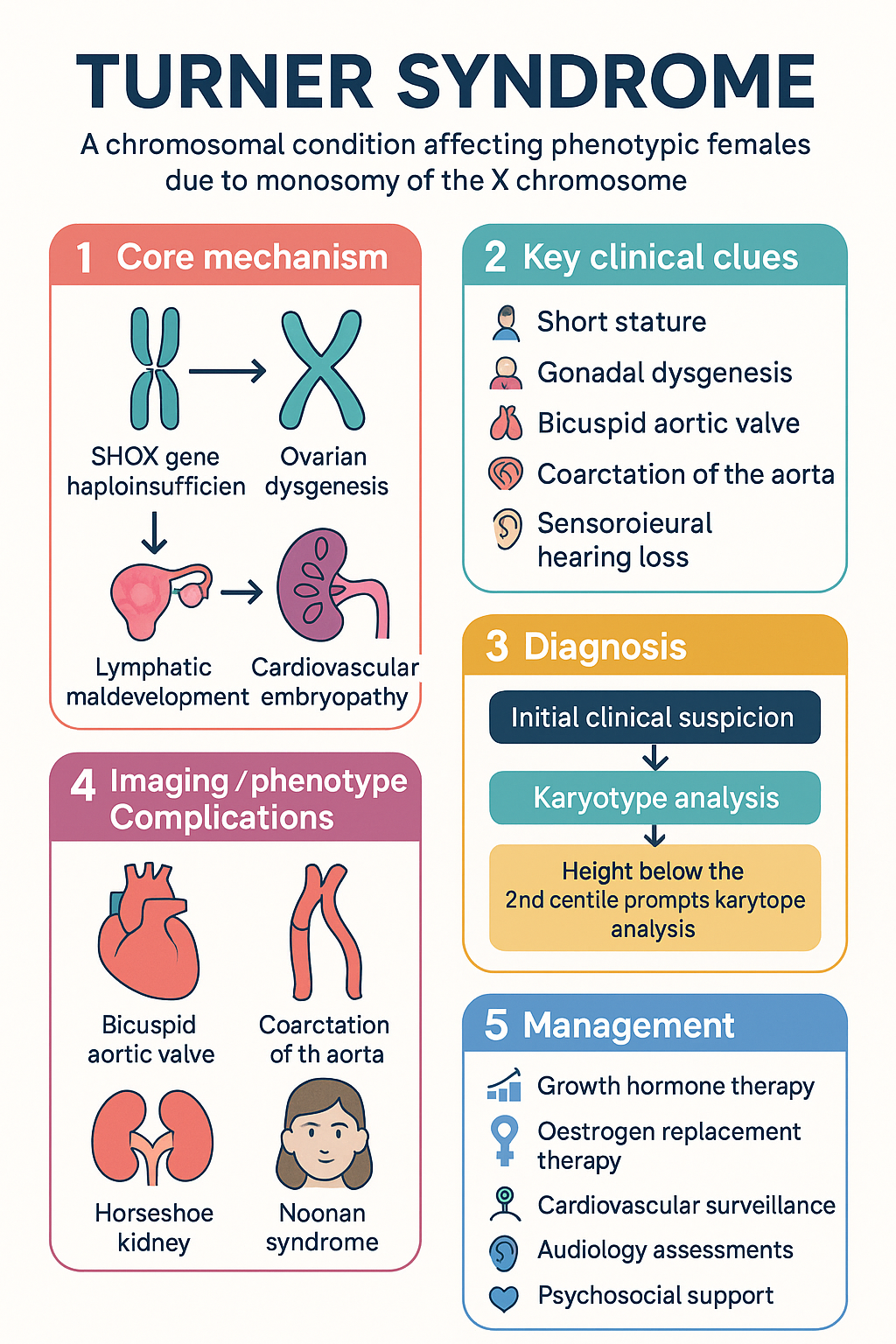
- Turner syndrome (TS) affects approximately 1 in 2500 live-born females and is characterised by complete or partial loss of the second X chromosome.
- Karyotype is the gold standard for diagnosis; mosaicism (45,X/46,XX) occurs in ~30% of cases and may present later with milder features.
- Short stature is near-universal and responsive to recombinant growth hormone (somatropin), which can add 5–8 cm to final adult height when commenced in early childhood.
- Gonadal dysgenesis with streak ovaries leads to hypergonadotropic hypogonadism in >90%; oestrogen replacement therapy (ORT) is essential from age 11–12 years to induce puberty and maintain bone health.
- Bicuspid aortic valve (BAV) occurs in ~30% and coarctation of the aorta in ~10%; lifelong cardiac surveillance every 5–10 years is mandatory.
- Aortic dissection risk is 6× general female population; blood pressure must be controlled to <130/80 mmHg and competitive sport restrictions may apply if aortic root dilation >2.5 cm/m² is present.
- Autoimmune thyroid disease (Hashimoto's thyroiditis) occurs in ~30%; annual TSH and anti-TPO antibodies from age 4 years is recommended.
- Renal anomalies (horseshoe kidney, duplex collecting system) are present in ~30% and warrant baseline renal ultrasound.
- Sensorineural hearing loss develops in up to 70% by adulthood; audiology assessment every 3–5 years is required.
- Fertility is possible in ~2% with spontaneous puberty; oocyte cryopreservation and IVF with donor oocytes are options but carry high-risk pregnancy considerations including aortic dissection.
- Transition of care from paediatric to adult endocrinology should occur by age 16–18 with a structured multidisciplinary plan.
- Psychosocial support, educational assessment for non-verbal learning difficulties, and screening for anxiety and depression are integral to holistic management.
🎧 Audio Brief
Introduction & Australian Epidemiology
Turner syndrome (TS) is a chromosomal condition affecting phenotypic females, resulting from complete or partial monosomy of the X chromosome. First described by Henry Turner in 1938, it is one of the most common chromosomal abnormalities in females and carries significant implications across multiple organ systems throughout the lifespan.
In Australia, TS occurs in approximately 1 in 2500 live-born female infants, equating to roughly 10–15 new diagnoses per year nationally. However, the true prevalence may be higher as up to 99% of 45,X conceptions are lost spontaneously in utero, and mosaicism (45,X/46,XX) can present subtly in adolescence or adulthood. The Australian Institute of Health and Welfare (AIHW) records TS as part of congenital anomaly surveillance, and the condition receives ongoing attention through state-based genetics services and paediatric endocrinology networks.
TS requires lifelong, multidisciplinary care spanning paediatric and adult endocrinology, cardiology, audiology, reproductive medicine, psychology, and renal medicine. Early diagnosis enables timely intervention for growth, puberty induction, and cardiovascular risk mitigation. This guideline provides an evidence-based framework for the diagnosis and management of TS in the Australian healthcare setting.
Genetics & Pathophysiology
Chromosomal Basis
TS arises from the loss of all or part of one X chromosome in phenotypic females. The karyotypic spectrum includes:
| Karyotype | Frequency | Clinical Notes |
|---|---|---|
| 45,X (complete monosomy) | ~45% | Most severe phenotype; usually diagnosed prenatally or in infancy |
| 45,X/46,XX (mosaicism) | ~30% | Variable phenotype; may have spontaneous puberty; diagnosed later |
| 46,X,i(Xq) | ~15% | Isochromosome Xq; risk of autoimmune disease and hearing loss |
| 46,X,del(Xp) | ~5% | Short stature gene (SHOX) lost from Xp; milder gonadal failure |
| 46,X,r(X) | ~5% | Ring chromosome; phenotype depends on XIST expression |
Pathophysiological Mechanisms
- SHOX haploinsufficiency: The SHOX gene (Short Stature Homeobox) on Xp22.33 escapes X-inactivation. Loss of one copy leads to impaired long-bone growth, accounting for the short stature characteristic of TS. This is the primary target for growth hormone therapy.
- Ovarian dysgenesis: Accelerated oocyte atresia results in streak ovaries by childhood or adolescence in most patients with 45,X. This leads to oestrogen and progesterone deficiency, absent puberty, and primary amenorrhoea.
- Lymphatic maldevelopment: Impaired lymphatic drainage in utero underpins nuchal cystic hygroma, peripheral lymphoedema, and contributes to left-sided cardiac anomalies including coarctation and BAV.
- Cardiovascular embryopathy: Abnormal endocardial cushion development results in BAV (~30%), coarctation of the aorta (~10%), and aortic root dilation predisposing to dissection.
- Epigenetic effects: Imprinting and X-inactivation patterns in mosaic individuals influence phenotypic variability. Skewed X-inactivation may modulate severity.
Clinical Features
The clinical presentation of TS varies by age, karyotype, and degree of mosaicism. Features may be detected antenatally, at birth, in childhood, or in adolescence/adulthood.
Antenatal & Neonatal Features
- Increased nuchal translucency or cystic hygroma on antenatal ultrasound
- Left-sided cardiac anomaly (coarctation, hypoplastic left heart) on foetal echocardiography
- Non-immune hydrops foetalis
- Low birth weight and short length for gestational age
- Peripheral lymphoedema of hands and feet (puffy hands/feet)
- Webbed neck (pterygium colli) from resolving cystic hygroma
- Low posterior hairline
- Shield chest with widely spaced hypoplastic nipples
Childhood Features
- Short stature — most common presenting feature; height velocity falls below the 25th centile from age 2–3 years
- Cubitus valgus (increased carrying angle)
- Short fourth metacarpal
- Multiple pigmented naevi
- Nail dysplasia (hyperconvex nails)
- Recurrent otitis media (common in early childhood)
- Feeding difficulties and failure to thrive in infancy
Adolescent & Adult Features
- Delayed puberty or primary amenorrhoea (absent breast development by age 13)
- Primary ovarian insufficiency (hypergonadotropic hypogonadism)
- Sensorineural hearing loss (high-frequency; progressive from age ~10 years)
- Autoimmune thyroiditis (hypothyroidism most common)
- Coeliac disease (prevalence ~4–6%)
- Hypertension (often from coarctation or essential)
- Aortic root dilation and risk of dissection
- Osteoporosis (oestrogen-dependent bone mineral accrual impaired)
- Non-verbal learning difficulties, executive function impairment
- Anxiety and depression (increased prevalence)
- Hepatic steatosis and abnormal LFTs
- Metabolic syndrome and type 2 diabetes mellitus (increased risk)
Features by Organ System
| System | Feature | Prevalence |
|---|---|---|
| Cardiovascular | Bicuspid aortic valve | ~30% |
| Coarctation of the aorta | ~10% | |
| Aortic root dilation | ~20–30% | |
| Renal | Horseshoe kidney | ~10% |
| Duplex collecting system | ~15% | |
| Endocrine | Hashimoto's thyroiditis | ~30% |
| Coeliac disease | 4–6% | |
| ENT/Audiology | Sensorineural hearing loss | up to 70% (adults) |
| Skeletal | Osteoporosis | ~25–50% |
| Psychosocial | Non-verbal learning difficulty | ~30–40% |
Investigations & Diagnosis
Diagnostic Approach
Diagnosis requires a high index of clinical suspicion. Karyotype analysis on peripheral blood lymphocytes (≥30 cells counted) is the gold standard. Diagnosis may be made prenatally (via amniocentesis or CVS for abnormal ultrasound findings), at birth, in childhood (short stature), or in adolescence (delayed puberty).
Baseline Investigations at Diagnosis
Differential Diagnosis
- Constitutional short stature / familial short stature
- Growth hormone deficiency (isolated)
- Noonan syndrome (46,XX or 46,XY; autosomal dominant; PTPN11 mutation)
- SHOX-related short stature (Léri-Weill dyschondrosteosis)
- 46,XY gonadal dysgenesis (Swyer syndrome) — if streak gonads but normal height
- Pure gonadal dysgenesis (46,XX)
Management
Management of TS is lifelong and multidisciplinary. Core pillars include growth hormone therapy, pubertal induction, cardiovascular surveillance, management of associated autoimmune and metabolic conditions, and psychosocial support.
1. Growth Hormone Therapy
Recombinant human growth hormone (somatropin) is the standard of care for short stature in TS and is PBS-subsidised under the Life Saving Drugs Program (LSDP) or as Authority Required for this indication.
2. Pubertal Induction & Oestrogen Replacement
Oestrogen replacement therapy (ORT) is essential for inducing secondary sexual characteristics, promoting uterine growth, achieving peak bone mass, and optimising quality of life. Pubertal induction should commence at age 11–12 years (or when clinically appropriate to match peers).
3. Cardiovascular Surveillance & Management
Cardiovascular disease is the leading cause of premature mortality in TS, primarily from aortic dissection and ischaemic heart disease. A structured surveillance programme is essential.
Blood pressure management: Target <130/80 mmHg. ACE inhibitors (enalapril, perindopril) or angiotensin receptor blockers (losartan) are preferred agents, particularly if aortic dilation is present. Beta-blockers may be added if needed.
4. Thyroid Disease Management
Hashimoto's thyroiditis is the most common autoimmune condition in TS (~30%). Annual TSH and fT4 monitoring from age 4 years is recommended. If hypothyroidism develops:
5. Fertility & Reproductive Counselling
Approximately 2–5% of women with TS achieve spontaneous conception, predominantly those with mosaic karyotypes who undergo spontaneous puberty. Fertility options include:
- Spontaneous conception: Possible in mosaic patients. Pregnancy must be managed in a tertiary centre due to aortic dissection risk (1–2% per pregnancy).
- Oocyte cryopreservation: May be considered in adolescents/adults with evidence of ovarian reserve (antral follicle count, AMH). Requires IVF referral.
- IVF with donor oocytes: Most common route to pregnancy in TS. Requires oestrogen priming of the uterus.
- Surrogacy: Legal option in some Australian states (varies by jurisdiction).
6. Bone Health
Osteoporosis risk is increased due to oestrogen deficiency, SHOX haploinsufficiency, and possibly intrinsic bone abnormalities. Management includes:
- Adequate calcium intake (1000–1300 mg/day) and vitamin D supplementation (1000–2000 IU/day if 25-OH vitamin D <75 nmol/L)
- Lifelong oestrogen replacement to maintain bone mineral density
- Weight-bearing exercise
- DXA scan at age 18–20 and then every 2–5 years, or earlier if oestrogen non-compliance
- Consider bisphosphonates (alendronate) if established osteoporosis (T-score <−2.5) — PBS Authority Required
7. Coeliac Disease
Screen with tTG-IgA and total IgA at diagnosis and periodically or if symptomatic (diarrhoea, iron deficiency, growth failure). Positive serology warrants confirmatory upper GI endoscopy with duodenal biopsy. Gluten-free diet is the treatment.
8. Hearing
Sensorineural hearing loss (SNHL) is progressive and primarily affects high frequencies (2000–8000 Hz). Conductive loss from chronic otitis media is common in childhood. Management includes:
- Audiology assessment every 3–5 years from age 4 years (annually if abnormal)
- Prompt treatment of otitis media; consider grommets if recurrent
- Hearing aids for SNHL
- Avoid ototoxic medications where possible (aminoglycosides)
9. Metabolic & Hepatic Surveillance
- Annual fasting glucose and/or HbA1c from age 10 years — type 2 diabetes risk is increased
- Annual LFTs — hepatic steatosis and elevated GGT/ALP are common
- Lipid profile every 2–5 years from age 10
- Promote healthy weight, physical activity, and Mediterranean-style diet
10. Psychosocial & Neurocognitive Support
Women and girls with TS often have normal verbal IQ but may experience specific difficulties with non-verbal processing, executive function, visuospatial skills, and mathematics. Psychosocial challenges include:
- Formal neuropsychological assessment at school entry and during adolescence
- Educational support plans (Individual Education Plans) if learning difficulties identified
- Screening for anxiety and depression at every clinic visit (PHQ-A, GAD-7)
- Peer support groups (e.g., Turner Syndrome Association of Australia)
- Counselling regarding body image, short stature, delayed puberty, and fertility
- Social work referral for complex psychosocial needs
Monitoring
Lifelong surveillance is essential. The following schedule is recommended (adapted from international TS consensus guidelines):
| Investigation | Frequency | Notes |
|---|---|---|
| Height/weight/BMI | Every 3–6 months (paediatric) | Plot on TS-specific growth charts |
| Thyroid function (TSH, fT4) | Annually from age 4 | More frequently if initiating GH or on thyroxine |
| Coeliac serology | At diagnosis, then if symptomatic or q2–3 years | tTG-IgA + total IgA |
| Echocardiography / Cardiac MRI | Every 5 years (low risk); 1–2 years (high risk) | MRI preferred from age 12 |
| Blood pressure | Every clinic visit | Target <130/80 mmHg |
| Audiology | Every 3–5 years (annually if abnormal) | Pure tone audiometry |
| LFTs | Annually | GGT, ALP, ALT commonly elevated |
| Fasting glucose / HbA1c | Annually from age 10 | Increased type 2 diabetes risk |
| Lipid profile | Every 2–5 years from age 10 | Cardiovascular risk stratification |
| DXA scan | At age 18–20, then every 2–5 years | Earlier if oestrogen non-adherence |
| IGF-1 (during GH therapy) | Annually | Target <2 SD above age-adjusted mean |
| Renal ultrasound | At diagnosis; repeat if abnormality found | Screen for structural anomalies |
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1–G70.
- 2. Saenger P, Wikland KA, Conway GS, et al. Recommendations for the diagnosis and management of Turner syndrome. J Clin Endocrinol Metab. 2001;86(7):3061–3069.
- 3. Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab. 2010;95(4):1487–1495.
- 4. Fiot E, Zenaty D, Boizeau P, et al. X-chromosome gene dosage as a determinant of congenital malformations and of age-related comorbidity risk in patients with Turner syndrome. Eur J Endocrinol. 2020;182(6):565–575.
- 5. Mavinkurve-Groothuis AMC, van der Putten K, van Kimmenade R, et al. Cardiac screening in women with Turner syndrome: a systematic review. Int J Cardiol. 2019;278:303–309.
- 6. Australian Institute of Health and Welfare (AIHW). Congenital anomalies in Australia. Cat. no. PER 102. Canberra: AIHW; 2022.
- 7. Royal Australasian College of Physicians (RACP). Transition care guidelines for adolescents with chronic conditions. Sydney: RACP; 2020.
- 8. Shankar RK, Backeljauw PF. Current best practice in the management of Turner syndrome. Ther Adv Endocrinol Metab. 2023;14:20420188231155368.
- 9. National Health and Medical Research Council (NHMRC). Australian guidelines for the clinical care of people with chronic kidney disease. Canberra: NHMRC; 2022.
- 10. Heart Foundation of Australia. Guidelines for the management of absolute cardiovascular disease risk. Melbourne: NHF; 2023.
- 11. Donaldson MDC, Gault EJ, Tan KW, Dunger DB. Optimising management in Turner syndrome: from infancy to adult transfer. Arch Dis Child. 2006;91(6):513–520.
- 12. The Australasian Society for the Study of Bone and Mineral Metabolism (ASBMR). Position statement on the management of osteoporosis in Australia. Sydney: ASBMR; 2022.