📋 Key Information Summary
- Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders affecting adrenal steroid biosynthesis, most commonly due to 21-hydroxylase deficiency (CYP21A2) — accounting for approximately 90–95% of all cases.
- Australia includes CAH in newborn bloodspot screening (NBS) programmes across all states and territories, enabling early detection of classical salt-wasting and simple-virilising forms.
- Classical CAH presents in the neonatal period with ambiguous genitalia (46,XX) or salt-wasting crisis (both sexes); non-classical CAH presents later with precocious puberty, acne, hirsutism, or infertility.
- Salt-wasting crisis is a medical emergency requiring IV 0.9% sodium chloride, IV hydrocortisone bolus (50–100 mg/m² in neonates), and dextrose; delayed treatment can be fatal.
- Lifelong glucocorticoid replacement with hydrocortisone is the cornerstone of management in children; modified-release hydrocortisone (Plenadren®) or prednisolone may be used in adults.
- Mineralocorticoid replacement with fludrocortisone is required for salt-wasting and simple-virilising classical CAH; sodium chloride supplementation is added in neonates and infants.
- Monitoring includes serum 17-hydroxyprogesterone (17-OHP), androstenedione, renin activity, growth velocity, and bone age in paediatric patients.
- Antimicrobial prophylaxis is not routinely required; stress dosing of hydrocortisone (2–3× maintenance) is essential during febrile illness, surgery, or trauma.
- Genetic counselling is recommended for all families; pre-conception carrier testing with CYP21A2 genotyping is available through Australian genetics services.
- Surgical management of ambiguous genitalia (feminising genitoplasty) should be performed at specialised centres with multidisciplinary DSD (differences of sex development) teams; timing remains individualised.
- Fertility is reduced in both sexes; assisted reproductive technologies and targeted ovulation induction can improve outcomes.
- ATSI populations may face barriers to specialist follow-up and access to genetic counselling, particularly in remote communities; active outreach is essential.
🎧 Audio Brief
Introduction & Australian Epidemiology
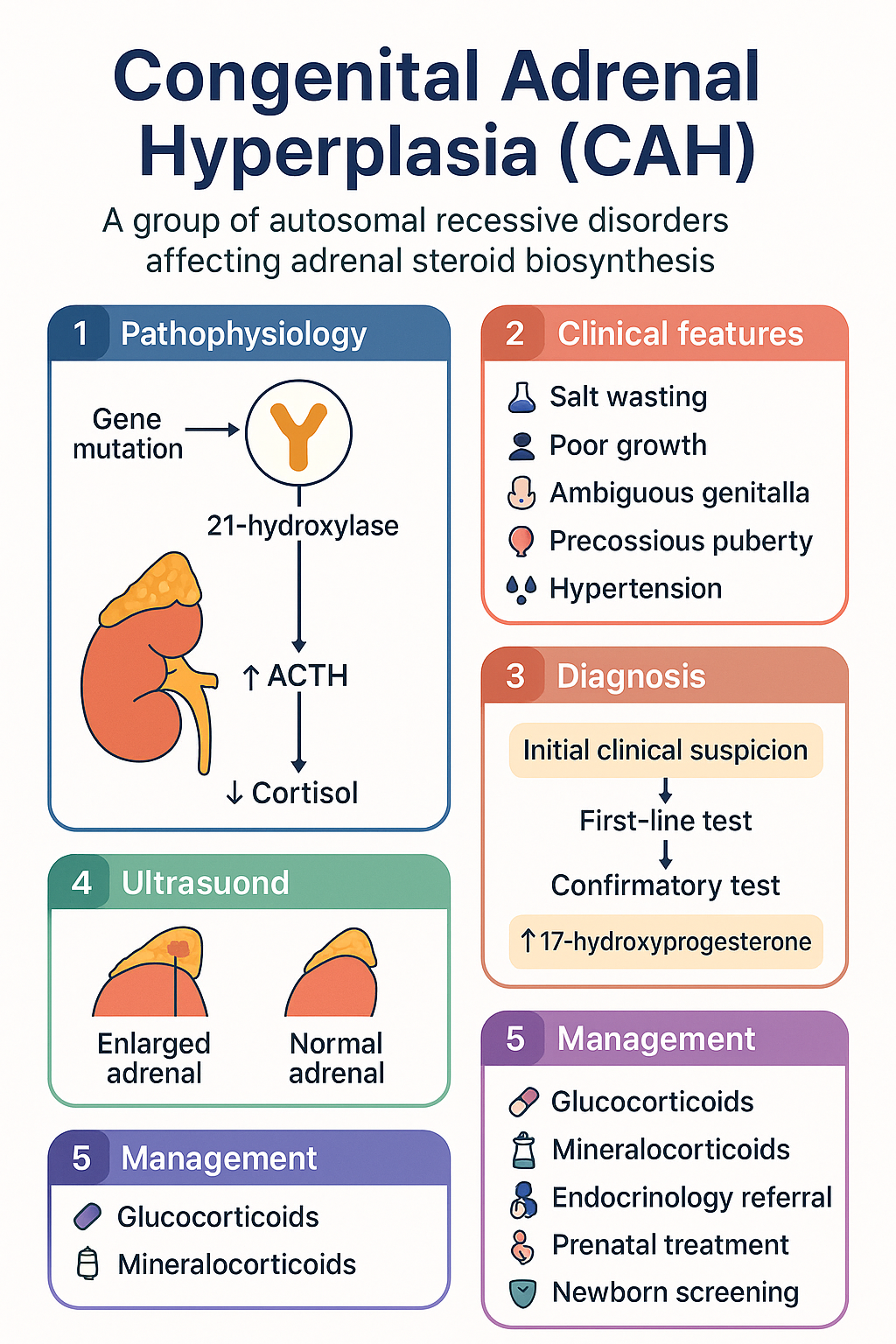
Congenital adrenal hyperplasia (CAH) comprises a family of autosomal recessive disorders characterised by impaired adrenal steroid biosynthesis. The condition results from deficiency of one of several enzymes required for the synthesis of cortisol, and in some cases aldosterone, from cholesterol. The most prevalent form — 21-hydroxylase deficiency due to mutations in the CYP21A2 gene — accounts for approximately 90–95% of all CAH cases worldwide and is the focus of this guideline.
The reported incidence of classical CAH (salt-wasting and simple-virilising forms) is approximately 1 in 15,000 live births globally. In Australia, newborn bloodspot screening (NBS) programmes have been operational in all states and territories since the early 2000s and reliably detect classical 21-hydroxylase deficiency. Data from the Australian Institute of Health and Welfare (AIHW) and state-based newborn screening laboratories suggest an incidence consistent with international figures, with no significant geographic variation within Australia.
Non-classical (late-onset) CAH is substantially more common, with prevalence estimates ranging from 1 in 200 to 1 in 1,000 depending on population. It is not detected by NBS and is typically diagnosed during adolescence or adulthood following investigation for hyperandrogenism, oligomenorrhoea, or infertility.
CAH management requires a multidisciplinary team including paediatric endocrinology, adult endocrinology, urology/gynaecology, clinical genetics, psychology, and nursing specialists. Early diagnosis, appropriate hormone replacement, and lifelong monitoring are critical to preventing adrenal crises, optimising growth and pubertal development, and preserving fertility.
Pathophysiology & Enzyme Defects
Adrenal steroidogenesis converts cholesterol through a series of enzymatic steps into cortisol, aldosterone, and adrenal androgens. Deficiency of any enzyme in this pathway leads to accumulation of precursors proximal to the block and shunting of intermediates into alternative steroid pathways. The resulting hormonal imbalance — cortisol deficiency with variable aldosterone deficiency and androgen excess — drives the clinical phenotype of CAH.
21-Hydroxylase Deficiency (CYP21A2)
21-hydroxylase (P450c21) catalyses the conversion of 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol (glucocorticoid pathway) and progesterone to 11-deoxycorticosterone (mineralocorticoid pathway). Deficiency results in:
- Impaired cortisol synthesis → loss of negative feedback → elevated ACTH → adrenal hyperplasia
- Accumulation of 17-OHP and progesterone → shunting to androgen pathway → excess DHEA, androstenedione, testosterone
- Impaired aldosterone synthesis (in salt-wasting form) → hyponatraemia, hyperkalaemia, dehydration
Other Enzyme Deficiencies
| Enzyme | Gene | Frequency | Key Features |
|---|---|---|---|
| 21-Hydroxylase | CYP21A2 | ~90–95% | Virilisation, salt wasting |
| 11β-Hydroxylase | CYP11B1 | ~5–8% | Virilisation, hypertension (DOC excess) |
| 3β-Hydroxysteroid dehydrogenase | HSD3B2 | ~1–2% | Mild virilisation (XX), undervirilisation (XY) |
| 17α-Hydroxylase / 17,20-lyase | CYP17A1 | <1% | Hypertension, sexual infantilism, 46,XY DSD |
| P450 oxidoreductase | POR | Rare | Antley–Bixler syndrome, skeletal anomalies |
| StAR / Cholesterol side-chain cleavage | STAR / CYP11A1 | Rare | Lipoid CAH — severe, all steroid production impaired |
CYP21A2 Genotype–Phenotype Correlation
Over 100 CYP21A2 mutations have been characterised. Genotype generally predicts phenotype severity:
- null / large deletions: No enzyme activity → salt-wasting CAH
- IVS2 splice mutation (intron 2 splice site): ~1–2% residual activity → salt-wasting CAH
- I172N (exon 4): ~2–5% residual activity → simple virilising CAH
- V281L (exon 7): ~20–50% residual activity → non-classical CAH
- P30L (exon 1): ~30–60% residual activity → non-classical CAH
Genotyping is available through Australian clinical genetics services (e.g., Victorian Clinical Genetics Services, SA Pathology Genetics, PathWest) and is recommended for confirmation of diagnosis, carrier testing, and prenatal counselling.
Classical vs Non-Classical CAH
Clinical Features & Newborn Screening
Neonatal Presentation
Classical CAH may present in the newborn period with:
- Ambiguous genitalia in 46,XX neonates: Ranges from clitoromegaly (Prader I) to complete labioscrotal fusion with penile urethra (Prader V). The degree of virilisation does not correlate with the degree of salt wasting.
- Salt-wasting crisis (both sexes): Typically onset at 1–3 weeks of life. Features include poor feeding, vomiting, lethargy, failure to thrive, dehydration, hypotension, hyponatraemia, hyperkalaemia, and metabolic acidosis. Can rapidly progress to cardiovascular collapse and death if untreated.
- Hyperpigmentation: Excess ACTH stimulates melanocortin receptors, causing generalised skin darkening, particularly of the scrotum/labia, areolae, and skin creases.
- 46,XY neonates with classical salt-wasting CAH appear phenotypically normal at birth and may not be diagnosed until the salt-wasting crisis.
Newborn Bloodspot Screening in Australia
All Australian states and territories include CAH (21-hydroxylase deficiency) in their newborn bloodspot screening programmes. The screening test measures immunoreactive 17-OHP from a heel-prick blood sample collected at 48–72 hours of life.
Childhood & Adolescent Presentation
- Simple virilising CAH diagnosed after the neonatal period may present with early pubic hair, body odour, clitoromegaly, penile enlargement, accelerated linear growth, and advanced bone age.
- Untreated patients develop premature epiphyseal fusion and short adult stature despite tall childhood height.
- Non-classical CAH in children presents with premature adrenarche (pubic/axillary hair before age 8 in girls, 9 in boys) and mild virilisation.
Adult Presentation
- Women: hirsutism, acne, menstrual irregularity, anovulation, infertility, and clitoromegaly. Frequently misdiagnosed as PCOS.
- Men: may be asymptomatic or have testicular adrenal rest tumours (TART), reduced spermatogenesis, and infertility.
- Both sexes: risk of adrenal crisis during intercurrent illness, surgery, or trauma if not on adequate stress dosing.
Diagnostic Investigations
Management: Glucocorticoids & Mineralocorticoids
Principles of Treatment
- Replace deficient cortisol (and aldosterone where needed) to suppress excess ACTH and reduce adrenal androgen production.
- Avoid both under-replacement (persistent hyperandrogenism, virilisation, accelerated bone age) and over-replacement (iatrogenic Cushing syndrome, growth suppression, obesity, osteoporosis).
- Treatment is lifelong; abrupt cessation can precipitate adrenal crisis.
- Stress dosing is essential during intercurrent illness, surgery, and trauma.
Glucocorticoid Replacement
Emergency / Stress Dosing of Hydrocortisone
Monitoring Targets
| Parameter | Target | Frequency |
|---|---|---|
| Serum 17-OHP | 1–10 nmol/L (mid-morning, pre-dose) — slightly above normal. Over-suppressed → overtreated. | Every 3–6 months (paediatric); 6–12 months (adult) |
| Serum androstenedione | Age- and sex-appropriate normal range | Every 3–6 months |
| Serum testosterone | Age- and sex-appropriate normal range | Every 3–6 months |
| Plasma renin activity | Within age-appropriate normal range (indicates adequate fludrocortisone) | Every 3–6 months |
| Serum Na⁺ / K⁺ | Normal range | Every 3–6 months (more frequent in neonates) |
| Growth velocity | 50th–75th percentile velocity; height within genetic target range | Every 3–6 months (paediatric) |
| Bone age (left wrist X-ray) | Within 1 SD of chronologic age (not advanced >2 years) | Annually in children; as needed in adults |
| Blood pressure | Age-appropriate normal range (over-replacement of fludrocortisone → hypertension) | Every visit |
| BMI / weight | Maintain healthy BMI. Monitor for Cushingoid features (over-replacement) | Every visit |
| Testicular ultrasound (males) | Screen for testicular adrenal rest tumours (TART) | Annually from puberty |
Adrenal Crisis Emergency Card
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Speiser PW, Arlt W, Auchus RJ, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(11):4043–4088.
- 2. El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet. 2017;390(10108):2194–2210.
- 3. Joint LWPES/ESPE CAH Working Group. Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab. 2002;87(9):4048–4053.
- 4. Royal Australasian College of Physicians (RACP). Paediatric endocrinology guidelines — Congenital adrenal hyperplasia. Sydney: RACP; 2020.
- 5. Australian Institute of Health and Welfare (AIHW). Newborn bloodspot screening national policy framework. Canberra: AIHW; 2018.
- 6. Falhammar H, Nyström HF, Ekman U, et al. Increased prevalence of cardiometabolic risk factors in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2023;108(3):e1009–e1018.
- 7. Whittle E, Falhammar H. Glucocorticoid regimens in the treatment of congenital adrenal hyperplasia: a systematic review and meta-analysis. J Endocr Soc. 2022;6(1):bvac165.
- 8. Pharmaceutical Benefits Scheme (PBS). Schedule of pharmaceutical benefits. Australian Government Department of Health. Available at: https://www.pbs.gov.au. Accessed 2024.
- 9. White PC. Neonatal screening for congenital adrenal hyperplasia. Nat Rev Endocrinol. 2009;5(9):490–498.
- 10. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, et al. Congenital adrenal hyperplasia — current insights in pathophysiology, diagnostics, and management. Endocr Rev. 2022;43(1):91–159.
- 11. Australasian Paediatric Endocrine Group (APEG). Clinical practice guidelines for management of congenital adrenal hyperplasia. Sydney: APEG; 2021.
- 12. Aboriginal and Torres Strait Islander Health Performance Framework 2020 summary report. Australian Institute of Health and Welfare (AIHW). Canberra: AIHW; 2020.