📋 Key Information Summary
- Sexual differentiation is a multi-step process involving chromosomal sex (XX/XY), gonadal sex, and phenotypic sex, each governed by distinct genetic and hormonal pathways.
- SRY on the Y chromosome is the master switch for testis determination; its absence leads to default ovarian development via pathways including WNT4 and FOXL2.
- Disorders of Sex Development (DSD) encompass congenital conditions in which chromosomal, gonadal, or anatomical sex is atypical — incidence approximately 1 in 4,500–5,500 live births.
- Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency is the most common cause of ambiguous genitalia in 46,XX individuals and constitutes a neonatal emergency if salt-wasting crises occur.
- All Australian states and territories screen for CAH via newborn bloodspot screening, enabling early detection and prevention of adrenal crisis.
- Klinefelter syndrome (47,XXY) is the most common sex chromosome aneuploidy in males, yet remains underdiagnosed — fewer than 25% of affected individuals receive a diagnosis.
- Turner syndrome (45,X and variants) affects approximately 1 in 2,500 live female births and requires lifelong surveillance for cardiovascular, endocrine, renal, and reproductive complications.
- Diagnosis of DSD requires a multidisciplinary team (MDT) approach including paediatric endocrinology, genetics, surgery, psychology, and social work.
- Karyotype, FISH, and targeted gene panels are first-line investigations; chromosomal microarray and whole-exome sequencing are increasingly used in complex or undiagnosed cases.
- Management of DSD centres on hormone replacement (cortisol/mineralocorticoids in CAH, testosterone in hypogonadal males, oestrogen in Turner syndrome), surgical intervention where indicated, and psychosocial support.
- Current Australian and international consensus emphasises a patient/family-centred, ethics-guided approach to gender assignment, deferring non-medically necessary genital surgery until the individual can participate in decision-making.
- Aboriginal and Torres Strait Islander communities face unique barriers to DSD diagnosis and care, including geographic remoteness, cultural considerations around gender and sexuality, and limited access to specialist paediatric endocrinology services.
🎧 Audio Brief
Introduction & Australian Epidemiology
Sexual differentiation refers to the sequential biological processes that transform a bipotential embryonic precursor into phenotypically male or female anatomy. The process operates across three levels — chromosomal sex, gonadal sex, and phenotypic sex — each governed by distinct but interacting genetic and endocrine pathways. Understanding these mechanisms is fundamental to diagnosing and managing Disorders of Sex Development (DSD), a group of congenital conditions in which chromosomal, gonadal, or anatomical sex is atypical.
In Australia, DSD collectively affects an estimated 1 in 4,500 to 5,500 live births, though milder variants (e.g., Klinefelter syndrome, Turner syndrome) may remain undiagnosed until adolescence or adulthood. Congenital adrenal hyperplasia (CAH) is the most common cause of ambiguous genitalia, with an incidence of approximately 1 in 15,000 in Australian populations. Newborn bloodspot screening programmes across all Australian states and territories now include CAH screening, significantly improving early detection and reducing morbidity from salt-wasting adrenal crises.
Australia's healthcare system mandates a multidisciplinary, family-centred approach to DSD care, consistent with the 2006 Chicago Consensus and subsequent updates from the DSD-TRN (Disorders of Sex Development — Translational Research Network) and the Australasian Paediatric Endocrine Group (APEG). Ethical considerations — including the timing of surgical intervention, gender assignment, and long-term psychosocial outcomes — are central to contemporary Australian practice.
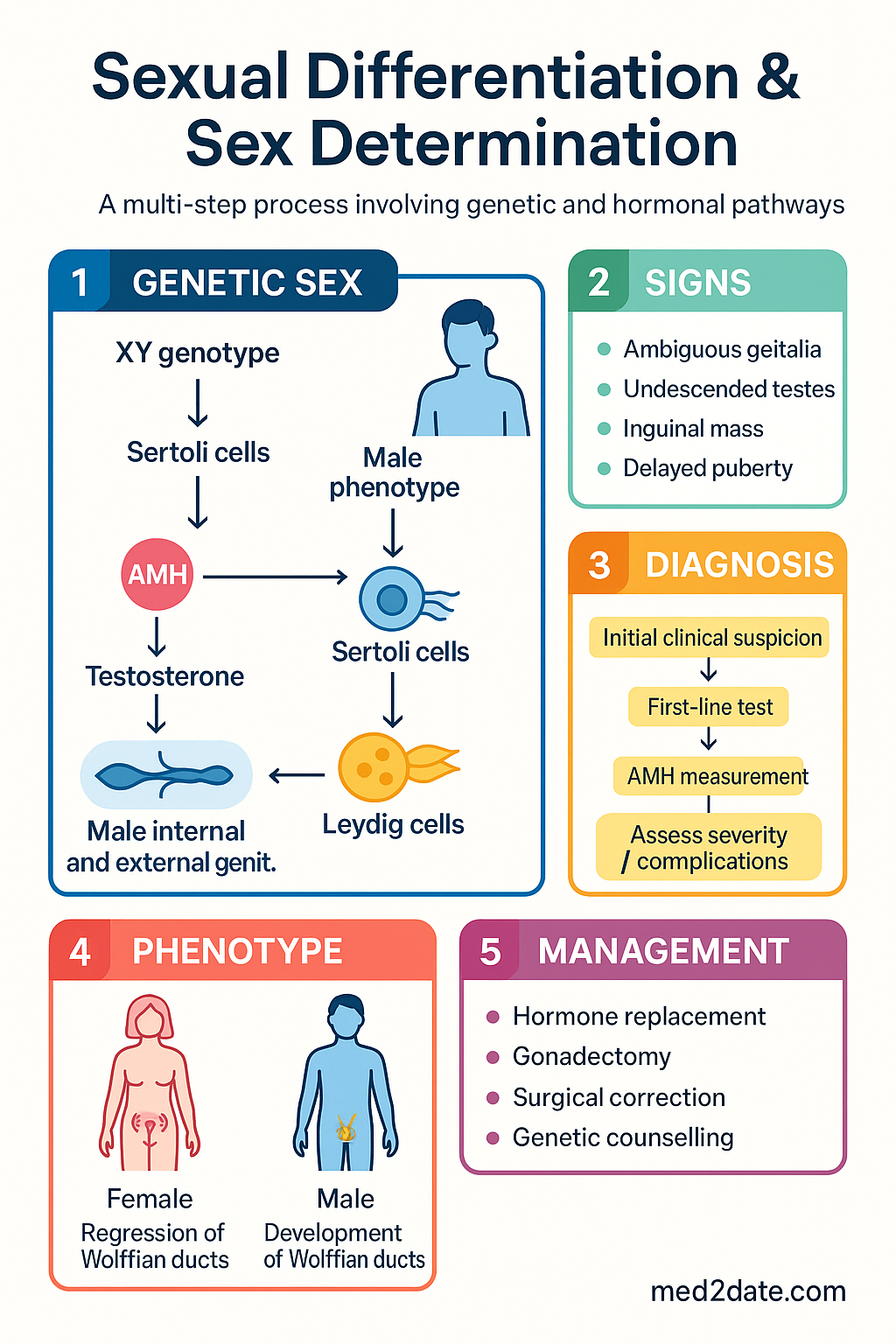
Embryology & Genetics
Bipotential Gonad
Until approximately the sixth week of gestation, the urogenital ridge is sexually undifferentiated. Primordial germ cells migrate from the yolk sac to the genital ridges, which contain coelomic epithelium, mesenchymal cells, and the mesonephric (Wolffian) and paramesonephric (Müllerian) ducts. The transcription factors SF1 (steroidogenic factor 1), WT1 (Wilms tumour 1), and LHX9 are essential for initial gonad formation.
Testis Determination Pathway
The presence of the SRY (sex-determining region Y) gene on the short arm of the Y chromosome (Yp11.3) triggers a cascade leading to Sertoli cell differentiation. SRY upregulates SOX9, which — together with SF1 and FGF9 — drives Sertoli cell commitment and testis cord formation. Sertoli cells then secrete two key hormones:
- Anti-Müllerian hormone (AMH) — causes regression of the Müllerian ducts (paramesonephric ducts) between weeks 8–10 of gestation.
- Testosterone (from Leydig cells, stimulated by placental hCG and later fetal LH) — stabilises the Wolffian ducts to form the epididymis, vas deferens, and seminal vesicles.
Testosterone is converted to dihydrotestosterone (DHT) by 5α-reductase type 2 in target tissues, driving external genital virilisation (penile urethra, scrotum, prostate).
Ovary Determination Pathway
In the absence of SRY, the default pathway favours ovarian development. WNT4 and RSPO1 signalling promotes β-catenin activation, which inhibits SOX9 and supports granulosa cell differentiation. FOXL2 maintains ovarian identity postnatally and actively suppresses testicular gene expression. Without testosterone and AMH, the Wolffian ducts regress and the Müllerian ducts develop into the fallopian tubes, uterus, and upper vagina.
Summary: Key Genes in Sex Determination
| Gene | Locus | Function | Loss-of-Function Phenotype |
|---|---|---|---|
| SRY | Yp11.3 | Testis-determining factor | 46,XY complete gonadal dysgenesis (Swyer syndrome) |
| SOX9 | 17q24.3 | Sertoli cell differentiation | 46,XY DSD; campomelic dysplasia |
| SF1 (NR5A1) | 9q33 | Gonadal & adrenal development | 46,XY DSD; adrenal insufficiency |
| WT1 | 11p13 | Gonadal & kidney development | Denys–Drash syndrome; Frasier syndrome |
| WNT4 | 1p36 | Ovarian pathway / Müllerian maintenance | 46,XX testicular/ovotesticular DSD |
| FOXL2 | 3q22.3 | Granulosa cell differentiation | Blepharophimosis–ptosis–epicanthus inversus syndrome (BPES) |
| SRD5A2 | 2p23.1 | 5α-reductase type 2 (T → DHT) | 5α-reductase deficiency; ambiguous genitalia at birth |
| AR | Xq12 | Androgen receptor | Complete androgen insensitivity syndrome (CAIS) |
| CYP21A2 | 6p21.33 | 21-hydroxylase (cortisol & aldosterone synthesis) | CAH — virilisation of 46,XX; salt-wasting crisis |
| AMH / AMHR2 | 19p13.3 / 12q13 | Müllerian duct regression | Persistent Müllerian duct syndrome (PMDS) |
Disorders of Sex Development (DSD)
The 2006 Chicago Consensus replaced older terms (intersex, pseudohermaphroditism, true hermaphroditism) with a classification based on karyotype. This nomenclature is endorsed by the Australasian Paediatric Endocrine Group (APEG) and is used throughout Australian clinical practice.
DSD Classification (Chicago 2006)
| Category | Examples | Key Features |
|---|---|---|
| Sex chromosome DSD | 47,XXY (Klinefelter); 45,X (Turner); 45,X/46,XY mosaicism; 46,XX/46,XY chimerism | Abnormal sex chromosome complement; variable gonadal and phenotypic outcomes |
| 46,XY DSD | Gonadal dysgenesis (partial/complete); 5α-reductase deficiency; 17β-HSD deficiency; CAIS; PAIS; LH receptor mutations | Y chromosome present but testis determination or androgen action impaired |
| 46,XX DSD | CAH (21-hydroxylase, 11β-hydroxylase, 3β-HSD); ovotesticular DSD; 46,XX testicular DSD (SRY translocation) | Y chromosome absent but virilisation occurs (endogenous or exogenous androgen exposure) |
Sex Chromosome DSD
Congenital Adrenal Hyperplasia (CAH)
CAH due to 21-hydroxylase deficiency (CYP21A2 mutations) accounts for over 90% of all CAH cases and is the most common cause of ambiguous genitalia in 46,XX newborns. Neonatal screening is performed in all Australian states via the Guthrie card bloodspot.
46,XY DSD — Androgen Insensitivity Syndrome (AIS)
AIS results from mutations in the androgen receptor (AR) gene on Xq12. Testes produce testosterone and AMH normally, so Müllerian structures are absent, but androgen action is impaired to varying degrees:
| Type | Phenotype | Presentation |
|---|---|---|
| Complete AIS (CAIS) | Female external phenotype; blind-ending vagina; testes intra-abdominal or inguinal | Primary amenorrhoea in phenotypic female; inguinal hernia in childhood |
| Partial AIS (PAIS) | Ambiguous genitalia; variable Wolffian development | Ambiguous genitalia at birth; gender assignment complex |
| Mild AIS (MAIS) | Male external phenotype; possible gynaecomastia, infertility | Infertility workup in adult males |
Clinical Features & Diagnosis
When to Suspect DSD
- Ambiguous genitalia at birth — clitoromegaly, labioscrotal fusion, micropenis, undescended testes, bifid scrotum
- Apparent female genitalia with inguinal hernia (consider CAIS)
- Apparent male genitalia with bilateral cryptorchidism or hypospadias with undescended testis
- Family history of DSD, consanguinity, neonatal death, or unexplained genital anomalies
- Primary amenorrhoea in phenotypic female with absent secondary sexual characteristics (consider Turner, CAIS)
- Virilisation in a 46,XX child (non-classical CAH, exogenous androgen exposure)
Diagnostic Approach
Investigations — Australian Availability
Management
Principles of Management
Australian management of DSD is guided by the following principles:
- Patient/family-centred care: The child and family are at the centre of all decisions. Information must be provided in a culturally appropriate, transparent, and supportive manner.
- Avoid irreversible interventions: Current Australian and international consensus recommends deferring non-medically necessary genital surgery (e.g., clitoroplasty, vaginoplasty, gonadectomy) until the individual can participate in decision-making, unless there is an urgent medical indication.
- MDT approach: All decisions regarding sex of rearing, hormone therapy, and surgery should be made by a DSD MDT with input from paediatric endocrinology, surgery, genetics, psychology, social work, ethics, and — when appropriate — adult endocrinology and reproductive medicine.
- Genetic counselling: Essential for all families, with discussion of recurrence risk, inheritance patterns, and implications for siblings.
- Psychosocial support: Ongoing psychological support for the child and family is a cornerstone of care. Gender identity and psychosocial well-being should be assessed regularly through childhood and adolescence.
Hormone Replacement — CAH
Hormone Replacement — Turner Syndrome
Surgical Management
Surgical decision-making in DSD is an area of active ethical debate. The following principles are endorsed by APEG and Australian paediatric surgical societies:
- Medically necessary surgery (e.g., surgery to relieve urinary obstruction, orchiopexy for intra-abdominal testes at risk of torsion, excision of dysgenetic gonads with malignancy risk) may be performed at any age when indicated.
- Elective feminising or masculinising genital surgery (e.g., clitoroplasty, vaginoplasty, hypospadias repair) — current consensus recommends deferring to adolescence where possible, allowing the patient to participate in decision-making. Family preferences and cultural context are considered by the MDT.
- Gonadectomy: Dysgenetic gonads (e.g., streak gonads in Swyer syndrome, intra-abdominal testes in CAIS) carry a risk of gonadoblastoma/dysgerminoma. Timing of prophylactic gonadectomy should be discussed with the MDT and family, balancing malignancy risk against the need for endogenous hormone production during puberty.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Lee PA, Houk CP, Ahmed SF, Hughes IA. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Archives of Disease in Childhood. 2006;91(7):554–563. doi:10.1136/adc.2006.098319
- 2. Hiort O, Birnbaum W, Marshall L, et al. Management of disorders of sex development. Nature Reviews Endocrinology. 2014;10(9):520–529. doi:10.1038/nrendo.2014.108
- 3. Speiser PW, Arlt W, Auchus RJ, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society Clinical Practice Guideline. Journal of Clinical Endocrinology & Metabolism. 2018;103(11):4043–4088. doi:10.1210/jc.2018-01865
- 4. Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome. European Journal of Endocrinology. 2017;177(3):G1–G70. doi:10.1530/EJE-17-0430
- 5. Bonomi M, Rochira V, Pasquali D, Balercia G, Jannini EA, Ferlin A. Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism. Journal of Endocrinological Investigation. 2017;40(2):123–134. doi:10.1007/s40618-016-0541-6
- 6. Australasian Paediatric Endocrine Group (APEG). Disorders of Sex Development: Clinical Practice Guidelines for Australian Paediatricians. APEG Position Statement. 2020. Available from: https://www.apeg.org.au
- 7. Intersex Human Rights Australia (IHRA). Intersex: Stories and Statistics from Australia. Melbourne: IHRA; 2021.
- 8. Cools M, Nordenström A, Robeva R, et al. Caring for individuals with a difference of sex development (DSD): A Consensus Statement. Nature Reviews Endocrinology. 2018;14(7):415–429. doi:10.1038/s41574-018-0010-8
- 9. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2020 summary report. Canberra: AIHW; 2020. Cat. no. IHW 248.
- 10. Royal Australasian College of Physicians (RACP). Standards for the care of children and adolescents with disorders of sex development: A consensus statement. Sydney: RACP; 2016.
- 11. Davies JH, Barton JS, Preece MA, Cameron-Pimblett A, Davies KM, Butler G. The transition from paediatric to adult endocrine care for patients with Turner syndrome. Clinical Endocrinology. 2020;92(5):393–401. doi:10.1111/cen.14163
- 12. National Health and Medical Research Council (NHMRC). National Statement on Ethical Conduct in Human Research 2023 (updated). Canberra: NHMRC; 2023.