📋 Key Information Summary
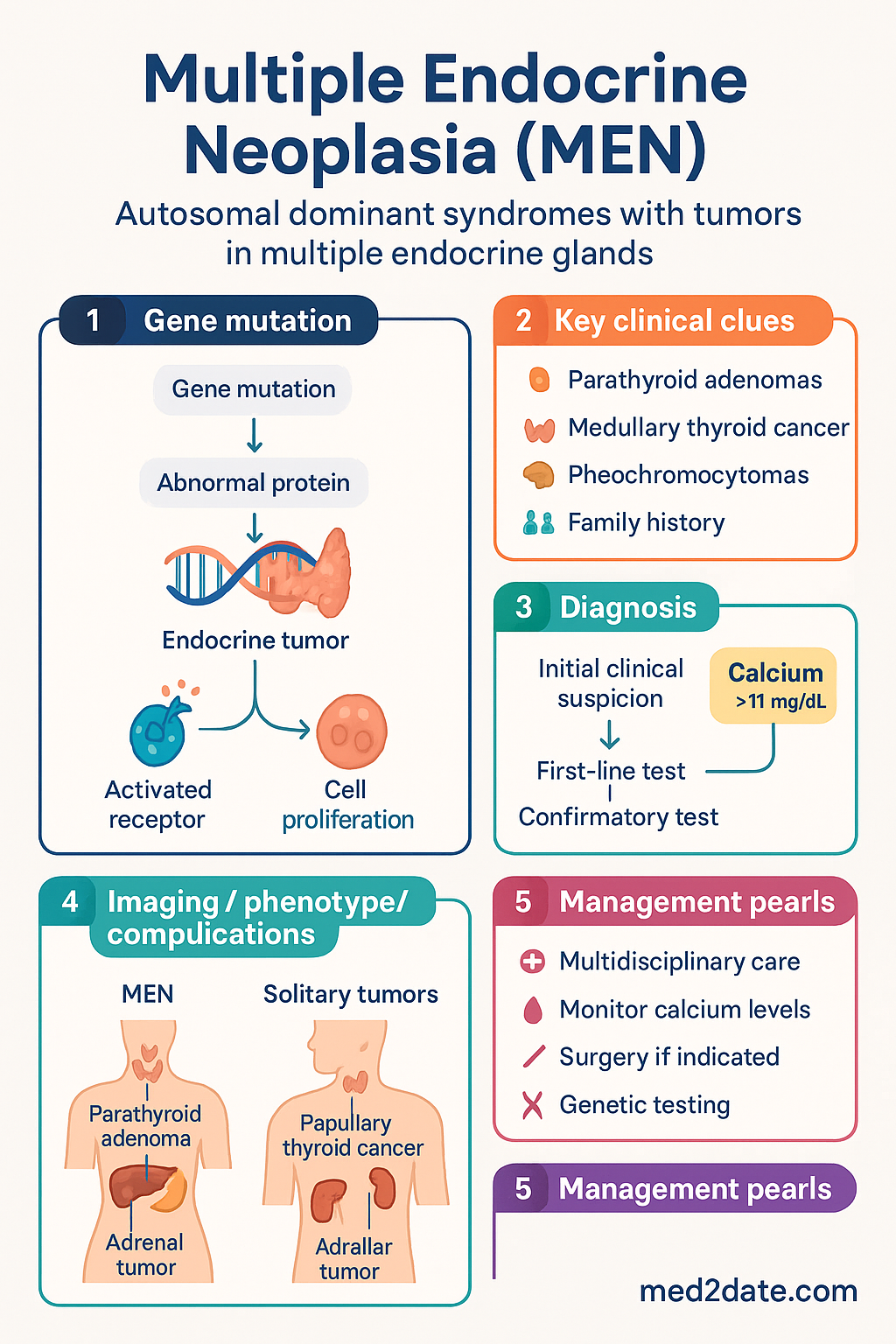
- Multiple Endocrine Neoplasia (MEN) syndromes are autosomal dominant conditions characterised by tumours in two or more endocrine glands; MEN1, MEN2A, MEN2B, and MEN4 are recognised subtypes.
- MEN1 (Wermer syndrome) is caused by pathogenic variants in the MEN1 tumour suppressor gene (11q13); penetrance >95% by age 50. Classic triad: primary hyperparathyroidism (≥90%), pancreatic neuroendocrine tumours (30–80%), and pituitary adenomas (15–50%).
- MEN2A (Sipple syndrome) is caused by activating mutations in the RET proto-oncogene (10q11.2); medullary thyroid carcinoma (MTC) in >95%, phaeochromocytoma ~50%, primary hyperparathyroidism 20–30%.
- MEN2B is caused by M918T RET mutation in ~95% of cases; most aggressive MTC phenotype with onset in infancy; also features mucosal neuromas, marfanoid habitus, and phaeochromocytoma.
- MEN4 is caused by CDKN1B pathogenic variants; phenocopies MEN1 with hyperparathyroidism and pituitary adenomas but lacks MEN1 mutations.
- Prophylactic thyroidectomy is the single most important intervention in MEN2: by age 6 months for MEN2B (highest risk), by age 5 years for MEN2A codon 634 mutations, and can be deferred with annual calcitonin surveillance for lower-risk MEN2A genotypes.
- Genetic testing with RET sequencing (MEN2) or MEN1/CDKN1B sequencing (MEN1/MEN4) should be offered to all first-degree relatives of mutation carriers.
- Rule out phaeochromocytoma before ANY surgery in MEN2A/2B patients — undiagnosed phaeochromocytoma during anaesthesia can cause fatal hypertensive crisis.
- Lifelong surveillance is mandatory in all MEN syndromes; refer to a MEN multidisciplinary clinic or endocrine genetics service where available.
- Aboriginal and Torres Strait Islander peoples may have reduced access to genetic testing and endocrine specialist services, particularly in remote areas; proactive outreach and culturally safe referral pathways are essential.
- Australian centres of excellence include the Royal Adelaide Hospital Endocrine Surgery Unit, Westmead Hospital Endocrine Genetics Clinic, and Peter MacCallum Cancer Centre genetics service.
- MBS item 73291 covers targeted RET mutation analysis; MBS item 73293 covers next-generation sequencing panels for hereditary cancer syndromes.
🎧 Audio Brief
Introduction & Australian Epidemiology
Multiple Endocrine Neoplasia (MEN) syndromes are a group of autosomal dominant inherited disorders that predispose affected individuals to the development of neoplasms in two or more endocrine glands. Four major subtypes are recognised: MEN1, MEN2A, MEN2B, and MEN4. These syndromes account for a significant proportion of apparently sporadic endocrine tumours and have important implications for family screening, surveillance, and prophylactic intervention.
The prevalence of MEN1 is estimated at 1 in 30,000 individuals, with no significant sex predilection. MEN2A is the most common MEN2 subtype (accounting for ~95% of MEN2 cases), with an estimated prevalence of 1 in 25,000–35,000. MEN2B is rarer (~5% of MEN2) but carries the most aggressive phenotype. MEN4 is the most recently described subtype and is considered very rare, with fewer than 100 families reported worldwide.
In Australia, the annual incidence of medullary thyroid carcinoma (MTC) — the hallmark of MEN2 — is approximately 0.14 per 100,000, with roughly 25–30% of MTC cases being hereditary (MEN2A or MEN2B). The AIHW Cancer Data Registry estimates that approximately 100–130 new cases of MTC are diagnosed annually, of which 25–40 are attributable to germline RET mutations. MEN1-related tumours are frequently encountered in Australian endocrine surgical practice, with parathyroidectomy being the most common initial surgical intervention.
| Feature | MEN1 | MEN2A | MEN2B | MEN4 |
|---|---|---|---|---|
| Gene | MEN1 (11q13) | RET (10q11.2) | RET (10q11.2) | CDKN1B (12p13) |
| Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant (~50% de novo) | Autosomal dominant |
| Prevalence | 1 : 30,000 | 1 : 25,000–35,000 | 1 : 400,000–1,000,000 | Very rare |
| Key tumours | Parathyroid, pituitary, pancreatic NET | MTC, phaeochromocytoma, parathyroid | MTC (aggressive), phaeochromocytoma, mucosal neuromas | Parathyroid, pituitary, other NET |
| Gene function | Tumour suppressor (menin) | Oncogene (receptor tyrosine kinase) | Oncogene (receptor tyrosine kinase) | Tumour suppressor (p27Kip1) |
MEN1 (Wermer Syndrome) — Parathyroid, Pituitary, Pancreas
Genetics & Pathophysiology
MEN1 is caused by inactivating germline mutations in the MEN1 tumour suppressor gene on chromosome 11q13, which encodes the nuclear protein menin. Menin interacts with JunD transcription factor, histone methyltransferases, and other regulators of cell proliferation. A somatic "second hit" in the remaining wild-type allele drives tumourigenesis according to the Knudson two-hit model. Over 1,300 distinct germline mutations have been described, with no clear genotype–phenotype correlation for most variants.
Approximately 10% of MEN1 cases arise from de novo germline mutations with no family history. Penetrance is age-dependent: by age 50, >95% of mutation carriers will have developed at least one MEN1-associated tumour.
Clinical Manifestations
Primary Hyperparathyroidism (≥90%)
The most common and usually earliest manifestation, presenting at a mean age of 20–25 years (decades earlier than sporadic primary hyperparathyroidism). Unlike sporadic disease, MEN1-related hyperparathyroidism involves multiple glands (multiglandular hyperplasia or asynchronous adenomas) in >95% of cases. Patients present with the classic triad of nephrolithiasis, bone loss, and non-specific symptoms (fatigue, depression, constipation, polydipsia).
Pancreatic Neuroendocrine Tumours (30–80%)
These are the leading cause of MEN1-related mortality. Tumours include:
- Gastrinomas (most common functional tumour, 40%): cause Zollinger–Ellison syndrome with refractory peptic ulcer disease and diarrhoea. Usually duodenal (often multiple and small).
- Insulinomas (10%): cause hypoglycaemia with Whipple triad. Usually benign.
- Non-functioning pancreatic NETs (30–50%): often detected on surveillance imaging; risk of malignancy increases with size >2 cm.
- Glucagonomas, VIPomas, somatostatinomas: rare but described.
Pituitary Adenomas (15–50%)
Prolactinomas are the most common subtype (~60% of MEN1 pituitary tumours), followed by non-functioning adenomas, GH-secreting adenomas (acromegaly), and ACTH-secreting adenomas (Cushing disease). MEN1-related pituitary adenomas tend to be larger and more aggressive than sporadic counterparts.
Other Associated Tumours
- Adrenocortical tumours (20–40%, usually non-functional)
- Thymic neuroendocrine tumours (2–8%, more common in males, aggressive)
- Bronchial carcinoid tumours (2–7%)
- Cutaneous lesions: facial angiofibromas, collagenomas, lipomas
Diagnosis
Clinical diagnosis of MEN1 requires the presence of tumours in at least two of the three principal MEN1-related endocrine organs (parathyroid, pituitary, pancreatic NETs). Familial MEN1 is defined as MEN1 in a patient with at least one first-degree relative with MEN1. Genetic confirmation by germline MEN1 mutation analysis is recommended in all suspected cases.
Investigations
Management
Hyperparathyroidism
Surgery is indicated when symptomatic or meeting standard operative criteria (serum calcium >0.25 mmol/L above upper limit of normal, eGFR <60 mL/min, T-score ≤ −2.5, age <50, nephrolithiasis/nephrocalcinosis). Subtotal parathyroidectomy (3.5-gland resection) with cervical thymectomy is the preferred initial approach. Total parathyroidectomy with autotransplantation (forearm) is an alternative. Recurrence rate is higher than in sporadic disease (~50% at 10 years).
Pancreatic NETs
Surgery is recommended for non-functioning pancreatic NETs >2 cm (or >1–2 cm with high-risk features) due to malignant potential. For functioning tumours (gastrinomas, insulinomas), surgery is indicated for symptom control and potential cure, though duodenal gastrinomas are often multifocal and may not be curable. Non-operative surveillance with 6–12 monthly imaging (CT/MRI/EUS) is appropriate for non-functioning tumours <1 cm.
Pituitary Adenomas
Management mirrors sporadic pituitary adenoma protocols. Dopamine agonists (cabergoline) are first-line for prolactinomas. Transsphenoidal surgery is indicated for non-functioning adenomas causing mass effect or hormone-secreting tumours unresponsive to medical therapy.
MEN2A (Sipple Syndrome) — Medullary Thyroid Carcinoma, Phaeochromocytoma, Hyperparathyroidism
Genetics & Pathophysiology
MEN2A is caused by activating germline mutations in the RET proto-oncogene on chromosome 10q11.2, which encodes a transmembrane receptor tyrosine kinase critical for neural crest cell development and differentiation. Over 95% of MEN2A patients harbour mutations in the extracellular cysteine-rich domain, most commonly at codon 634 (exon 11, ~85% of MEN2A). Other mutation sites include codons 609, 611, 618, 620 (exon 10), and codons 630, 768, 790, 791, 804, 891 (exons 13–15).
| ATA Risk Level | RET Codon | Recommended Thyroidectomy Timing |
|---|---|---|
| Highest | M918T (MEN2B) | Within 6 months of life |
| High | C634R/G/Y/W/S, A883F | By age 5 years (or earlier if calcitonin elevated) |
| Moderate | 609, 611, 618, 620, 768, 790, 791, 804, 891 | May defer if calcitonin normal; operate by age 5–10 or when calcitonin rises |
Clinical Manifestations
Medullary Thyroid Carcinoma (>95%)
MTC arises from parafollicular C cells and is virtually universal in MEN2A, typically presenting in the second to third decade (earlier than sporadic MTC). It is preceded by C-cell hyperplasia, a premalignant condition. Early MTC may be asymptomatic; advanced disease presents with a palpable thyroid nodule, cervical lymphadenopathy, or symptoms from metastatic disease (diarrhoea, flushing from calcitonin/serotonin secretion).
Phaeochromocytoma (~50%)
Bilateral in ~50% of affected patients. Phaeochromocytoma in MEN2A is almost always benign and typically secretes epinephrine (adrenaline) preferentially, in contrast to VHL-related phaeochromocytomas which secrete norepinephrine (noradrenaline). Presents with paroxysmal or sustained hypertension, headache, palpitations, diaphoresis, and anxiety. Can be life-threatening if undiagnosed prior to surgery.
Primary Hyperparathyroidism (20–30%)
Usually milder than in MEN1; typically multiglandular. Asymptomatic hypercalcaemia is common. Indications for parathyroidectomy are the same as for sporadic primary hyperparathyroidism.
Variant MEN2A Phenotypes
- MEN2A with cutaneous lichen amyloidosis (CLA): Associated with codon 634 mutations; pruritic skin lesion in the interscapular region.
- MEN2A with Hirschsprung disease: Associated with exon 10 mutations (codons 609, 611, 618, 620).
Investigations
Management
Prophylactic Total Thyroidectomy
The cornerstone of MEN2A management. Timing is dictated by ATA risk level (see table above). Total thyroidectomy with central lymph node dissection is standard. Patients require lifelong levothyroxine replacement post-operatively.
Phaeochromocytoma
Adrenalectomy is indicated for confirmed phaeochromocytoma. Cortical-sparing adrenalectomy is preferred in bilateral cases to avoid lifelong adrenal insufficiency; long-term steroid replacement and adrenal crisis education are required if total adrenalectomy is performed. Pre-operative alpha-blockade (phenoxybenzamine or doxazosin) for ≥10–14 days followed by beta-blockade if tachycardic is mandatory.
Advanced/Metastatic MTC
For progressive, unresectable, or metastatic MTC, tyrosine kinase inhibitors targeting RET are now available in Australia.
MEN2B & MEN4
MEN2B
Genetics
MEN2B is the most aggressive MEN2 subtype and is caused by the M918T mutation in exon 16 of RET in approximately 95% of cases. A rarer mutation, A883F (exon 15), accounts for most of the remainder. Approximately 50% of MEN2B cases arise from de novo germline mutations, meaning there is often no family history — a critical point for clinical suspicion.
Clinical Features
Management — MEN2B
Prophylactic thyroidectomy is urgent in MEN2B. The ATA 2020 guidelines recommend total thyroidectomy within the first 6 months of life, ideally before age 1 year. Central lymph node dissection should be performed concurrently if calcitonin is elevated. Delay beyond 1 year significantly worsens prognosis.
Post-operative surveillance includes serum calcitonin and CEA every 6 months for the first 5 years, then annually. Phaeochromocytoma screening (plasma metanephrines) should begin by age 11 years (or earlier if clinically suspected) and continue annually.
MEN4
Genetics & Phenotype
MEN4 is caused by heterozygous loss-of-function mutations in CDKN1B, encoding the cyclin-dependent kinase inhibitor p27Kip1. It was first described in 2006 and is sometimes termed "MEN1 phenocopy." The phenotype closely resembles MEN1: primary hyperparathyroidism is the most common manifestation (80%), followed by pituitary adenomas (40%), and neuroendocrine tumours of the pancreas, stomach, or lung. However, the clinical spectrum and penetrance are less well defined due to the rarity of reported cases.
Diagnosis & Management
MEN4 should be considered in patients with MEN1-like features who test negative for MEN1 mutations. Genetic testing for CDKN1B mutations is available through research laboratories and specialised genetic services (e.g., Peter MacCallum Cancer Centre, Genetic Health Queensland). Management follows MEN1 surveillance and treatment protocols, as no genotype-specific guidelines currently exist.
Genetic Testing & Surveillance
Who Should Be Tested?
- All first-degree relatives of known RET (MEN2) or MEN1 (MEN1) mutation carriers.
- Patients with apparently sporadic MTC (~25–30% harbour germline RET mutations).
- Patients with two or more MEN1-associated tumours.
- Patients with MTC diagnosed before age 35, bilateral MTC, or MTC with C-cell hyperplasia.
- Index cases testing negative for MEN1 should be considered for CDKN1B (MEN4), CDC73, and AIP testing.
Australian Genetic Testing Access
| Test | MBS Item | Provider Examples | Turnaround |
|---|---|---|---|
| Targeted RET mutation | 73291 | SA Pathology, VCGS, Douglass Hanly Moir | 2–4 weeks |
| NGS hereditary cancer panel | 73293 | Peter MacCallum, VCGS, NSW Health Pathology | 4–8 weeks |
| MEN1 sequencing | 73293 | SA Pathology, Genetic Health Queensland | 4–8 weeks |
Surveillance Protocols
Lifelong surveillance is essential. Below is a summary of recommended screening for RET and MEN1 mutation carriers.
MEN1 Mutation Carriers — Annual Screening
RET Mutation Carriers — ATA-Guided Surveillance
Genetic Counselling
All families with confirmed MEN mutations should be referred to a clinical geneticist or genetic counsellor for pre- and post-test counselling, discussion of at-risk relatives, implications for family planning (including preimplantation genetic testing, available at select Australian IVF centres), and psychosocial support. Contact your state genetic service (e.g., Genetic Health Queensland 1800 812 868, Victorian Clinical Genetics Services 03 8341 6200).
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Wells SA Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567–610.
- 2. American Thyroid Association Guidelines Working Group on Medullary Thyroid Carcinoma, et al. ATA 2020 guidelines update: management of medullary thyroid carcinoma. Thyroid. 2020.
- 3. Brandi ML, Agarwal SK, Eng C, et al. Multiple endocrine neoplasia type 1: latest insights. Endocr Rev. 2021;42(2):133–170.
- 4. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990–3011.
- 5. Frank-Raue K, Raue F. Hereditary medullary thyroid cancer genotype-phenotype correlation. Recent Results Cancer Res. 2015;204:139–156.
- 6. Machens A, Dralle H. Genotype-phenotype based surgical concept of hereditary medullary thyroid carcinoma. World J Surg. 2007;31(5):957–968.
- 7. Wohllk N, Schweizer H, Erlic Z, et al. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab. 2010;24(3):371–387.
- 8. Australian Institute of Health and Welfare (AIHW). Cancer data in Australia. Canberra: AIHW; 2024. Available from: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia
- 9. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. 2006;103(42):15558–15563.
- 10. National Health and Medical Research Council (NHMRC). Genetic testing for heritable mutations in human genes: guidance on the evaluation of genetic tests. Canberra: NHMRC; 2019.
- 11. Lenders JWM, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942.
- 12. Wirth LJ, Sherman E, Robinson B, et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N Engl J Med. 2020;383(9):825–835.
- 13. Royal Australasian College of Surgeons (RACS). Position statement on prophylactic thyroidectomy in MEN2. Melbourne: RACS; 2022.