📋 Key Information Summary
- Klinefelter's syndrome (47,XXY) is the most common sex chromosome aneuploidy in males, affecting approximately 1 in 600 live male births; Kallmann's syndrome (KS) is a form of congenital hypogonadotropic hypogonadism (HH) with olfactory impairment, prevalence ~1 in 30,000 males and ~1 in 120,000 females.
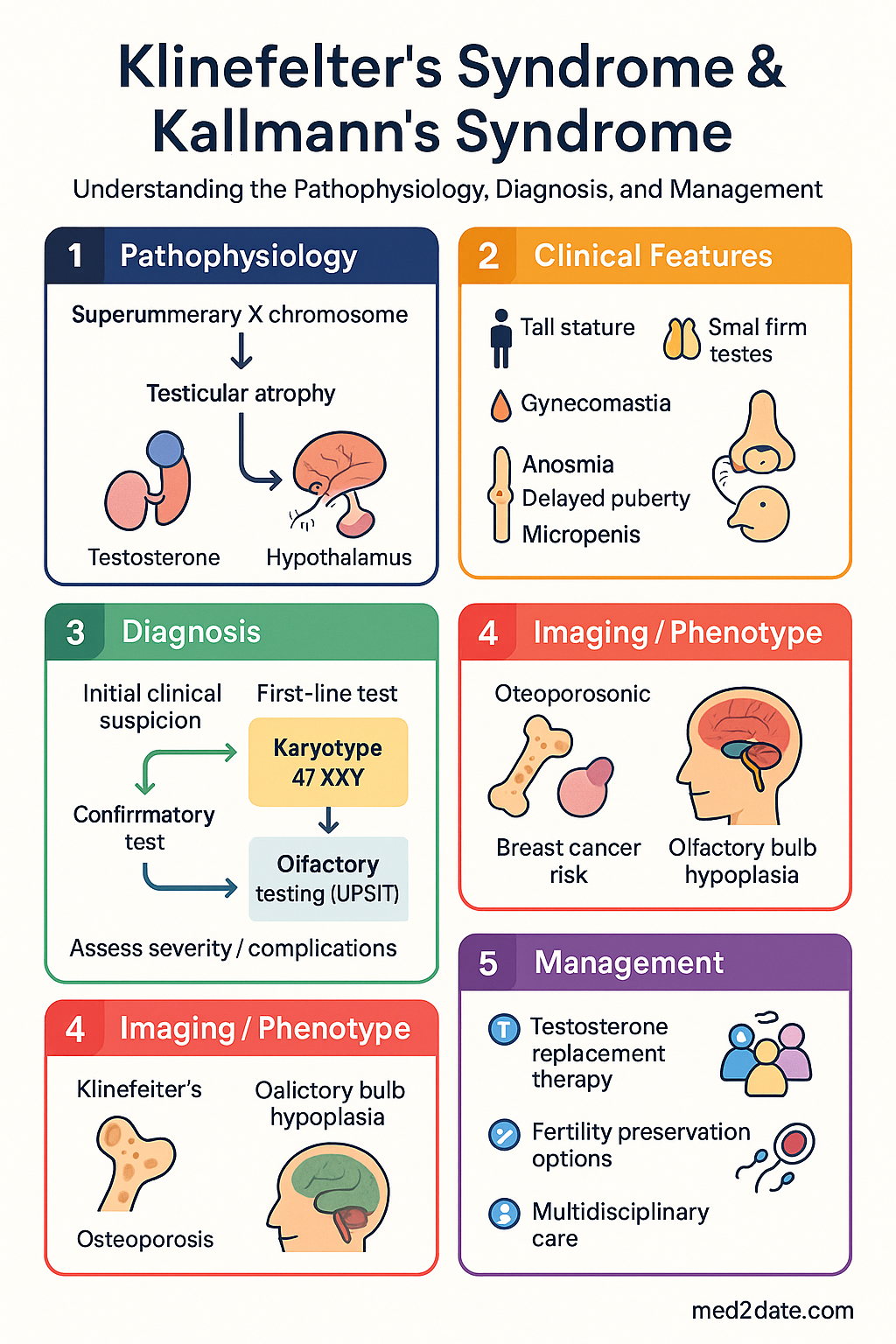
- Klinefelter's syndrome results from supernumerary X chromosome(s) leading to testicular fibrosis, small firm testes, hypergonadotropic hypogonadism, and infertility; 80–90% of affected individuals remain undiagnosed in Australia.
- Kallmann's syndrome arises from deficient GnRH neuronal migration; patients present with absent/delayed puberty and anosmia/hyposmia due to olfactory bulb hypoplasia.
- Both conditions require lifelong testosterone replacement therapy (TRT) — intramuscular testosterone undecanoate (Nebido®) or testosterone enanthate are the mainstay for adults in Australia.
- Klinefelter's — karyotype 47,XXY confirms diagnosis; testosterone is low or low-normal with elevated FSH/LH (hypergonadotropic pattern).
- Kallmann's — low testosterone with inappropriately low/normal FSH/LH (hypogonadotropic pattern); olfactory testing (UPSIT or Sniffin' Sticks) is diagnostic.
- Fertility preservation: Klinefelter's patients may harbour focal spermatogenesis — micro-TESE has 40–70% sperm retrieval rates; Kallmann's patients may respond to pulsatile GnRH or gonadotrophin therapy for fertility induction.
- Associated comorbidities in Klinefelter's include metabolic syndrome, type 2 diabetes, osteoporosis, venous thromboembolism, breast cancer (20–50× male baseline risk), and psychosocial difficulties.
- Kallmann's syndrome may feature associated midline defects (cleft palate, renal agenesis, synkinesia/mirror movements, hearing loss) and requires multi-disciplinary evaluation.
- Testosterone undecanoate (Nebido® 1000 mg IM every 10–14 weeks) is PBS-listed as Authority Required for established hypogonadism; testosterone enanthate (Primoteston® 250 mg IM every 2–3 weeks) is PBS General Benefit.
- Bone mineral density monitoring with DEXA is recommended from early adulthood in both conditions; vitamin D and calcium optimisation are essential.
- Psychological support and referral to clinical genetics, endocrinology, andrology, and mental health services should be offered at diagnosis and throughout life.
- Aboriginal and Torres Strait Islander males face additional barriers to diagnosis and ongoing care; culturally safe, community-based models improve outcomes.
🎧 Audio Brief
Introduction & Australian Epidemiology
Klinefelter's syndrome (47,XXY) and Kallmann's syndrome are two distinct yet clinically overlapping disorders of male hypogonadism that commonly present to primary care, paediatric endocrinology, and adult endocrinology services across Australia. Both conditions result in testosterone deficiency and impaired spermatogenesis, but their aetiologies, genetic architectures, and associated comorbidity profiles differ substantially.
Klinefelter's Syndrome
Klinefelter's syndrome is the most common sex chromosome aneuploidy in males, with an estimated prevalence of 1 in 600 live male births. In Australia, this translates to roughly 20,000–25,000 affected individuals, the vast majority of whom remain undiagnosed. Population-based screening data suggest that only 10–25% of cases are identified during the patient's lifetime. The condition was first described by Harry Klinefelter in 1942 and is characterised by the presence of one or more supernumerary X chromosomes in phenotypic males. Variants include 48,XXXY, 48,XXYY, 49,XXXXY, and mosaic patterns (46,XY/47,XXY), which collectively account for approximately 10–20% of Klinefelter karyotypes and tend to produce more severe phenotypes.
The Australian Institute of Health and Welfare (AIHW) does not maintain a specific register for Klinefelter's syndrome; however, extrapolation from European newborn screening programmes suggests that approximately 400 Australian males are born with 47,XXY each year. The condition contributes significantly to male infertility, accounting for approximately 3–5% of infertile men presenting to Australian andrology clinics.
Kallmann's Syndrome
Kallmann's syndrome (KS) is a rare form of congenital hypogonadotropic hypogonadism (CHH) combined with defective olfactory bulb development, resulting in anosmia or severe hyposmia. Prevalence estimates are 1 in 30,000 males and 1 in 120,000 females, with a male-to-female ratio of approximately 4:1. The condition is genetically heterogeneous, with autosomal dominant (ANOS1/KAL1, FGFR1), autosomal recessive (PROKR2, PROK2, CHD7), and oligogenic inheritance patterns described. Approximately 30–40% of cases harbour identifiable pathogenic variants, implying that additional gene discovery is ongoing.
In the Australian context, Kallmann's syndrome is managed through specialist endocrinology and andrology services in tertiary centres. The Royal Children's Hospital Melbourne, Westmead Children's Hospital Sydney, and the Queensland Children's Hospital all maintain paediatric endocrinology services capable of managing delayed puberty due to CHH. Adult transition care remains a critical gap, with many patients lost to follow-up during the transfer from paediatric to adult services.
Genetics & Pathophysiology
Klinefelter's Syndrome — Genetic Basis
The classic karyotype is 47,XXY, resulting from nondisjunction during meiosis I (approximately 50% paternal origin) or meiosis II (approximately 50% maternal origin). Approximately 10–20% of patients are mosaic (46,XY/47,XXY), which generally confers a milder phenotype with variable fertility potential. Higher-order aneuploidies (48,XXXY, 48,XXYY, 49,XXXXY) are rarer and associated with more pronounced intellectual disability, dysmorphic features, and multi-organ involvement.
The supernumerary X chromosome is subject to X-inactivation; however, escape genes — notably SHOX (Short Stature Homeobox) — contribute to the tall stature phenotype. The additional X chromosome leads to testicular dysgenesis through mechanisms that are not fully elucidated but include germ cell apoptosis, Sertoli cell dysfunction, and progressive hyalinisation and fibrosis of seminiferous tubules beginning in early childhood and accelerating at puberty.
Klinefelter's — Pathophysiology
The primary pathology in Klinefelter's syndrome is testicular failure:
- Sertoli cell dysfunction: Leads to elevated inhibin B loss of negative feedback, causing markedly elevated FSH (often >20 IU/L).
- Leydig cell impairment: Testosterone production is reduced or low-normal; compensatory LH elevation occurs (typically >10 IU/L). Leydig cell hyperplasia is observed histologically but is functionally insufficient.
- Germ cell depletion: Azoospermia is present in >90% of non-mosaic 47,XXY men; however, focal spermatogenesis may persist in mosaic individuals or be recoverable via micro-TESE.
- Metabolic consequences: Testosterone deficiency promotes insulin resistance, visceral adiposity, dyslipidaemia, and reduced bone mineral density.
Kallmann's Syndrome — Genetic Basis
Kallmann's syndrome results from defective migration of GnRH-secreting neurons from the olfactory placode to the hypothalamus during embryogenesis. The identified gene loci include:
| Gene | Inheritance | Frequency | Key Features |
|---|---|---|---|
| ANOS1 (KAL1) | X-linked recessive | 5–10% | Renal agenesis, synkinesia, more severe anosmia |
| FGFR1 | Autosomal dominant | ~10% | Cleft lip/palate, dental agenesis, digital anomalies |
| PROKR2 / PROK2 | Autosomal recessive | ~5–8% | Variable expressivity, fibrous dysplasia |
| CHD7 | Autosomal dominant | ~5–6% | CHARGE syndrome overlap, hearing loss, semicircular canal anomalies |
| FGF8 / FGF17 | Autosomal dominant | Rare | Oligogenic modifier, variable penetrance |
Kallmann's — Pathophysiology
The hallmark of Kallmann's syndrome is failed embryonic migration of GnRH neurons, which normally travel from the olfactory epithelium along olfactory nerve fibers through the cribriform plate to the medial basal hypothalamus. Concurrent failure of olfactory bulb and tract development produces the characteristic anosmia. The result is absent or insufficient pulsatile GnRH secretion, leading to:
- Low FSH and LH (hypogonadotropic pattern) despite low testosterone.
- Absent or incomplete puberty: micropenis, cryptorchidism, sparse/absent secondary sexual characteristics.
- Azoospermia or severe oligospermia (reversible with gonadotrophin therapy in many cases).
- Bone maturation delay and reduced peak bone mass if untreated.
Importantly, the hypothalamic–pituitary–gonadal axis is intact but unstimulated in Kallmann's syndrome; patients may therefore respond to exogenous GnRH (pulsatile) or gonadotrophins (hCG ± FSH), distinguishing them from primary gonadal failure seen in Klinefelter's syndrome.
Clinical Features
Klinefelter's Syndrome
The clinical presentation varies widely depending on age, karyotype, and degree of testosterone deficiency:
Kallmann's Syndrome
Clinical features reflect both the hypogonadotropic hypogonadism and associated developmental anomalies:
Differentiating Features
| Feature | Klinefelter's (47,XXY) | Kallmann's Syndrome |
|---|---|---|
| Gonadotrophins (FSH/LH) | Elevated (hypergonadotropic) | Low / inappropriately normal (hypogonadotropic) |
| Testosterone | Low or low-normal | Low |
| Olfactory function | Normal | Absent or markedly reduced |
| Stature | Tall (eunuchoid) | Normal or tall (if delayed epiphyseal fusion) |
| Testes | Small, firm (<6 mL) | Small, soft (prepubertal) |
| Gynaecomastia | Common (30–50%) | Uncommon |
| Fertility potential | Very low (micro-TESE possible) | Potentially recoverable with gonadotrophins |
| Karyotype | 47,XXY (or variant) | 46,XY (or 46,XX) |
Investigations & Diagnosis
Klinefelter's Syndrome
Kallmann's Syndrome
Management
Testosterone Replacement Therapy (TRT)
Lifelong testosterone replacement is the cornerstone of management for both Klinefelter's and Kallmann's syndromes. The goal is to restore physiological testosterone levels, promote and maintain virilisation, protect bone density, improve metabolic parameters, and enhance quality of life.
TRT Monitoring Protocol
Fertility Management
Fertility options differ substantially between the two conditions:
Klinefelter's Syndrome
- Micro-TESE: Microsurgical testicular sperm extraction achieves sperm retrieval in 40–70% of 47,XXY men. Retrieved sperm can be used for ICSI (intracytoplasmic sperm injection). Referral to a specialised andrologist/REI unit is recommended before age 35, as retrieval rates may decline with age.
- Sperm banking: If any sperm are found on ejaculate analysis (rare in non-mosaic Klinefelter's), cryopreservation should be offered.
- Pre-treatment counselling: TRT suppresses residual spermatogenesis — patients considering future fertility should undergo micro-TESE before commencing TRT, or TRT should be discontinued for 3–6 months prior to fertility attempts.
Kallmann's Syndrome
- Pulsatile GnRH therapy: Subcutaneous GnRH (gonadorelin) via portable pump, 75–200 ng/kg every 90–120 minutes. Physiological approach that stimulates endogenous FSH/LH secretion. Spermatogenesis achieved in ~80% of men. Not currently PBS-listed; sourced through hospital pharmacies.
- hCG ± FSH therapy: hCG (Ovidrel® 1500–2000 IU SC 2–3× weekly) mimics LH; add recombinant FSH (Gonal-F® 75–150 IU SC 3× weekly) if spermatogenesis does not progress after 6 months. PBS Authority Required for fertility indications.
- Expected timeline: Spermatogenesis typically achieved within 12–24 months of gonadotrophin therapy; sperm banking should be offered once sperm appear.
- Female partners: Women with Kallmann's syndrome (rarer) can undergo ovulation induction with pulsatile GnRH or gonadotrophins (FSH/hCG) — referral to reproductive endocrinology.
Management of Specific Comorbidities
- Gynaecomastia: Surgical (mastectomy) if persistent despite TRT; tamoxifen 10–20 mg PO daily may be trialled for 3–6 months (off-label, specialist-initiated).
- Metabolic syndrome: Lifestyle intervention, metformin if impaired glucose tolerance, statins per cardiovascular risk.
- Osteoporosis: Calcium 1000–1200 mg + vitamin D 1000 IU daily; bisphosphonates if T-score ≤ −2.5 or fragility fracture.
- VTE risk: Counsel regarding 2-fold increased VTE risk; consider thrombophilia screen before TRT in high-risk patients.
- Psychosocial: Refer to psychologist/psychiatrist; learning support for childhood diagnosis; support groups (e.g., Klinefelter's Syndrome Association of Australia).
- Bone health: More severely affected due to absent/delayed puberty; early TRT essential; calcium + vitamin D from diagnosis; DEXA annually until bone density stabilises.
- Renal anomalies: Screen with renal ultrasound; unilateral renal agenesis requires annual BP monitoring and eGFR.
- Hearing loss: Audiometry at diagnosis; hearing aids if indicated; early intervention for speech/language in paediatric patients.
- Psychosocial: Delayed puberty profoundly impacts adolescent identity and mental health; early psychological support is critical; transition planning for adult services.
- Cleft palate/dental: Multidisciplinary craniofacial team management; orthodontic review.
Pubertal Induction (Both Conditions)
Pubertal induction should commence at the age-appropriate time (typically 12–14 years) or when the patient is psychologically ready, using a graduated approach:
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Quick Reference — Key Management Algorithms
📚 References
- 1. Bonomi M, Vezzoli V, Krausz C, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol. 2018;178(1):23–32.
- 2. Gravholt CH, Chang S, Wallentin M, et al. Klinefelter syndrome: integrating genetics, neuropsychology, and endocrinology. Endocr Rev. 2018;39(4):389–423.
- 3. Radicioni AF, Ferlin A, Balercia G, et al. Consensus statement on diagnosis and clinical management of Klinefelter syndrome. J Endocrinol Invest. 2010;33(11):839–850.
- 4. Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism — pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11(9):547–564.
- 5. Franik S, Hoeijmakers Y, D'Hauwers K, et al. Klinefelter syndrome and fertility: sperm preservation should not be offered to children with Klinefelter syndrome. Hum Reprod. 2016;31(9):1953–1959.
- 6. Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice. 9th edn. East Melbourne: RACGP; 2016.
- 7. Australian Institute of Health and Welfare (AIHW). The health of Indigenous Australians. Cat. no. IHW 51. Canberra: AIHW; 2023.
- 8. Dwyer AA, Quinton R, Pitteloud N, et al. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: a systematic review and meta-analysis. Fertil Steril. 2015;104(5):1180–1188.
- 9. Franik S, Kremer JA, Nelen WE, et al. Aromatase inhibitors for subfertile men with elevated FSH. Cochrane Database Syst Rev. 2015;(9):CD010387.
- 10. Tournaye H, Krausz C, Oates RD. Concepts in diagnosis and therapy for male reproductive impairment. Lancet Diabetes Endocrinol. 2017;5(7):554–567.
- 11. Salonia A, Rastrelli G, Hackett G, et al. Paediatric and adult-onset male hypogonadism. Nat Rev Dis Primers. 2019;5(1):38.
- 12. Australian Government Department of Health. Pharmaceutical Benefits Schedule — testosterone undecanoate (Nebido). Canberra: PBS; 2024. Available from: www.pbs.gov.au.
- 13. Bry-Gauillard H, Larrat-Ledoux F, Maione L, et al. Congenital hypogonadotropic hypogonadism in women: clinical spectrum and evaluation. J Clin Endocrinol Metab. 2023;108(6):1482–1493.
- 14. Aboriginal and Torres Strait Islander Health Practice Board of Australia. Scope of practice guidelines. 2023. Available from: www.ahpra.gov.au.
- 15. Sánchez-Guijo A, Bentz NF, Wunsch R, et al. Metabolic syndrome in Klinefelter syndrome: impact of testosterone replacement. Eur J Endocrinol. 2020;183(2):187–197.