📋 Key Information Summary
- Acromegaly is caused by a GH-secreting pituitary adenoma in >95% of cases, leading to excess GH and IGF-1 with multi-system morbidity and increased mortality if untreated.
- Incidence in Australia is approximately 3–4 per million per year; prevalence is estimated at 60–70 per million, with typical diagnosis at age 40–50 years.
- Diagnostic delay averages 7–10 years due to insidious onset; raised serum IGF-1 (age- and sex-adjusted) is the initial screening test of choice.
- Oral glucose tolerance test (OGTT) with GH nadir ≥1 µg/L confirms diagnosis (dynamic suppression test); discordant results require specialist review.
- MRI pituitary with gadolinium is essential for tumour localisation and characterisation; microadenomas (<10 mm) are more amenable to surgical cure.
- Transsphenoidal surgery is first-line treatment for most patients, with biochemical remission rates of 80–90% for microadenomas and 40–60% for macroadenomas.
- Somatostatin analogues (octreotide LAR, lanreotide Autogel) are first-line medical therapy for residual or persistent disease post-surgery, normalising IGF-1 in 50–70% of patients.
- Pegvisomant, a GH receptor antagonist, is reserved for patients inadequately controlled on somatostatin analogues; normalises IGF-1 in >90% of cases (PBS Authority Required).
- Treatment targets are normalisation of age-adjusted IGF-1, random GH <1 µg/L, and GH nadir <0.4 µg/L on OGTT (ultrasensitive assay).
- Cardiovascular disease (cardiomyopathy, arrhythmias), diabetes mellitus, sleep apnoea, and colorectal neoplasia are major causes of excess morbidity and mortality.
- Multidisciplinary pituitary team management (endocrinologist, neurosurgeon, ophthalmologist, radiologist, pathologist) is mandatory for optimal outcomes.
- Lifelong surveillance is required after remission: annual IGF-1, 3-monthly post-surgery GH/IGF-1 for 5 years, then annual; monitor for hypopituitarism post-surgery.
🎧 Audio Brief
Introduction & Australian Epidemiology
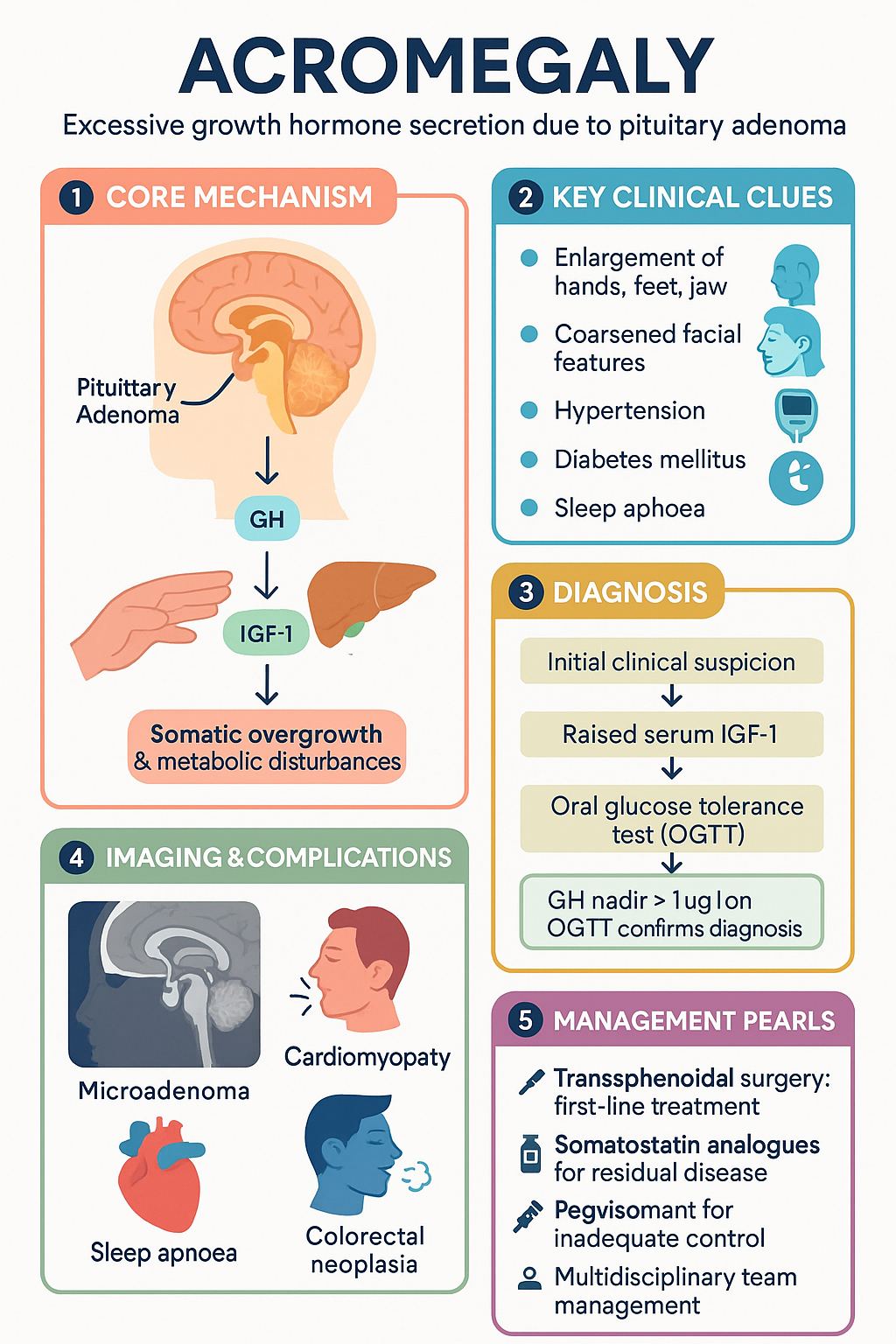
Acromegaly is a chronic, progressive endocrine disorder caused by excessive secretion of growth hormone (GH), almost always arising from a GH-secreting pituitary adenoma (somatotroph adenoma). The sustained hypersecretion of GH leads to overproduction of insulin-like growth factor 1 (IGF-1) from the liver and other tissues, resulting in characteristic somatic overgrowth, metabolic disturbances, and significant multi-system morbidity.
In Australia, the estimated annual incidence is 3–4 cases per million population, with a point prevalence of approximately 60–70 per million. These figures may underestimate the true burden, as diagnosis is typically delayed by 7–10 years owing to the insidious onset of clinical features. The condition is diagnosed most commonly in the fourth to fifth decades of life, with an equal sex distribution or slight female predominance in some series.
Untreated acromegaly is associated with a two- to three-fold increase in mortality, principally driven by cardiovascular disease, cerebrovascular events, and respiratory complications. Achievement of biochemical remission reduces mortality to that of the general population, underscoring the importance of early diagnosis and appropriate treatment. This guideline provides a comprehensive, Australian-contextualised approach to the diagnosis and management of acromegaly.
Pathophysiology & Epidemiology
Molecular Pathogenesis
The vast majority of GH-secreting pituitary adenomas are sporadic, arising from clonal expansion of a single mutated somatotroph cell. Key molecular mechanisms include:
- GNAS1 gene mutations: Activating somatic mutations in the GNAS1 gene (encoding the Gsα subunit) occur in approximately 40% of GH-secreting adenomas, leading to constitutive cAMP production and unregulated GH secretion. These are somatic mutations confined to the tumour.
- AIP mutations: Aryl hydrocarbon receptor interacting protein (AIP) germline mutations account for approximately 20% of familial isolated pituitary adenoma (FIPA) and are associated with younger-onset, larger, and more aggressive somatotroph adenomas. Carriers require genetic counselling and family screening.
- MEN1 and other syndromes: Multiple endocrine neoplasia type 1 (MEN1) causes pituitary adenomas in ~40% of carriers. Carney complex (PRKAR1A mutations) and McCune–Albright syndrome (GNAS1 mosaic mutations) are rarer familial causes of GH excess.
GH–IGF-1 Axis Physiology
GH is secreted by somatotroph cells of the anterior pituitary gland in a pulsatile fashion, regulated by hypothalamic growth hormone-releasing hormone (GHRH, stimulatory) and somatostatin (inhibitory). Ghrelin from the stomach also stimulates GH release. GH acts on the liver and peripheral tissues to stimulate IGF-1 production, which mediates most of the growth-promoting and metabolic effects. In acromegaly, autonomous GH secretion overrides normal feedback mechanisms, resulting in sustained elevation of both GH and IGF-1.
Tumour Characteristics
GH-secreting adenomas are classified as microadenomas (<10 mm diameter) or macroadenomas (≥10 mm). Macroadenomas are more common at presentation (60–70%) and are associated with extrasellar extension, compression of surrounding structures, and lower surgical remission rates. Invasive adenomas (Knosp grade 3–4), which encase the carotid artery, are present in approximately 25–35% of cases.
Australian Epidemiological Context
Data from the Australian Pituitary Registry and population-based studies indicate that acromegaly accounts for approximately 10–15% of all pituitary adenomas managed at major tertiary centres (e.g., Royal Melbourne Hospital, Westmead Hospital, Royal Adelaide Hospital). Aboriginal and Torres Strait Islander populations may face diagnostic delays due to healthcare access barriers, although specific prevalence data in this population are limited. Comorbidities at diagnosis in Australian cohorts include hypertension (40–50%), diabetes mellitus (15–38%), and sleep apnoea (60–70%).
Clinical Features
The clinical features of acromegaly develop gradually over years and are often attributed to ageing, contributing to the characteristic diagnostic delay. Features may be categorised by system:
Somatic / Musculoskeletal
- Enlargement of acral structures: hands (increased ring/glove size), feet (increased shoe size), jaw (prognathism, malocclusion), brow (frontal bossing)
- Soft tissue swelling leading to coarsened facial features, macroglossia, thickened skin
- Arthropathy — a major cause of morbidity; occurs in up to 70% of patients, involving weight-bearing joints initially with characteristic widened joint spaces, osteophyte formation, and eventual degenerative changes
- Carpal tunnel syndrome (50–60%), often bilateral
- Proximal myopathy with reduced exercise capacity
Cardiovascular
- Hypertension (40–50%) — multifactorial: sodium retention, endothelial dysfunction, sympathetic activation
- Acromegalic cardiomyopathy: concentric biventricular hypertrophy → diastolic dysfunction → systolic failure (in advanced cases); arrhythmias common
- Increased cardiovascular mortality is the leading cause of death in untreated acromegaly
Metabolic / Endocrine
- Insulin resistance → impaired glucose tolerance (16–46%) or overt diabetes mellitus (15–38%)
- Dyslipidaemia (hypertriglyceridaemia)
- Hypogonadism (from tumour mass effect or hyperprolactinaemia): 30–50% of men, menstrual irregularity in women
- Hypopituitarism from mass effect — GH, ACTH, TSH, gonadotrophin deficiency
Respiratory
- Obstructive sleep apnoea (60–70%): due to macroglossia, pharyngeal soft tissue hypertrophy, craniofacial changes
- Upper airway obstruction risk during anaesthesia
Neurological / Local Tumour Effects
- Headache (common, may be due to dural stretch or cavernous sinus invasion)
- Visual field defects (bitemporal hemianopia) from optic chiasm compression in macroadenomas
- Cranial nerve palsies (III, IV, VI) with lateral extension into the cavernous sinus
- Hyperprolactinaemia (30–40%) from co-secretion or stalk effect
Dermatological
- Skin tags (acrochordons) — associated with colonic polyps
- Hyperhidrosis, oily skin
- Acanthosis nigricans (associated with insulin resistance)
Neoplasia Risk
An increased risk of colorectal neoplasia (adenomas and possibly carcinoma) is well-documented; Australian guidelines recommend baseline colonoscopy at diagnosis and surveillance every 3–5 years, particularly in patients with additional risk factors (age >50, family history, skin tags). Thyroid nodules are also more prevalent and warrant ultrasound evaluation.
Investigations (IGF-1, Oral Glucose Tolerance Test)
Biochemical Diagnosis
IGF-1 (Insulin-like Growth Factor 1)
Serum IGF-1, measured by immunoassay and interpreted against age- and sex-adjusted reference ranges, is the initial screening investigation. IGF-1 reflects integrated 24-hour GH secretion and is elevated in virtually all patients with active acromegaly. A normal age-adjusted IGF-1 effectively excludes the diagnosis. IGF-1 is available through most Australian pathology services (MBS item 66658, Growth factors).
Oral Glucose Tolerance Test (OGTT) — Confirmatory Test
The 75 g OGTT is the gold-standard confirmatory test for acromegaly. In healthy individuals, glucose loading suppresses GH to nadir <0.4 µg/L (ultrasensitive assay) or <1 µg/L (standard assay). In acromegaly, autonomous GH secretion fails to suppress:
- GH nadir ≥0.4 µg/L (ultrasensitive assay) — diagnostic of acromegaly
- GH nadir ≥1 µg/L (standard assay) — diagnostic of acromegaly
- GH nadir 0.4–1.0 µg/L on ultrasensitive assay — equivocal; requires specialist interpretation with IGF-1 and clinical context
OGTT is not reliable in uncontrolled diabetes mellitus. The test should be performed fasting, in the morning, with GH measured at 0, 30, 60, 90, and 120 minutes. MBS item 66658 applies.
Random GH
A single random GH measurement is insufficient for diagnosis due to pulsatile secretion, but a random GH <0.4 µg/L (ultrasensitive assay) in the setting of elevated IGF-1 may indicate assay discordance or an alternative cause of IGF-1 elevation. Serial GH measurements or the OGTT are needed for definitive diagnosis.
Additional Baseline Investigations
Treatment Targets / Criteria for Remission
| Parameter | Normal / Remission Target | Notes |
|---|---|---|
| Age-adjusted IGF-1 | Within normal range for age and sex | Most important single marker of disease control |
| Random GH | <1 µg/L (standard) or <0.4 µg/L (ultrasensitive) | Reflects nadir during diurnal sampling |
| GH nadir on OGTT | <0.4 µg/L (ultrasensitive) | Best post-surgical prognostic marker |
| GH average on day curve | <1 µg/L | Useful for assessing medical therapy efficacy |
Management (Surgery, Somatostatin Analogues, Pegvisomant)
Management of acromegaly requires a multidisciplinary pituitary tumour board (endocrinologist, neurosurgeon, neuroradiologist, ophthalmologist, pathologist) and should be coordinated through an accredited pituitary centre of excellence. Treatment goals are biochemical remission, tumour control, relief of mass effect, and management of comorbidities.
1. Surgery — Transsphenoidal Adenomectomy
Transsphenoidal surgery (TSS) is the first-line treatment for the majority of patients with acromegaly and is the only potentially curative modality. Surgery should be performed by an experienced pituitary neurosurgeon (volume ≥50 transsphenoidal cases per year) at a major centre.
Post-operative considerations include assessment of pituitary function (particularly cortisol, thyroid function) at 6–12 weeks, MRI at 3 months, and biochemical remission assessment (IGF-1 ± OGTT) at 12 weeks post-surgery. Transient diabetes insipidus occurs in 10–20%, with permanent diabetes insipidus in <5% in experienced hands. CSF leak, meningitis, and carotid artery injury are rare but serious complications.
2. Somatostatin Analogues — First-Line Medical Therapy
Somatostatin receptor ligands (SRLs) are the first-line medical therapy for residual or recurrent acromegaly post-surgery, or as primary therapy when surgery is contraindicated or declined. They act on somatostatin receptors (predominantly SSTR2 and SSTR5) on the adenoma to suppress GH secretion.
3. Pegvisomant — GH Receptor Antagonist
Pegvisomant is a genetically engineered GH receptor antagonist that blocks peripheral GH action, thereby reducing IGF-1 production. It does not directly reduce tumour GH secretion and therefore does not shrink the adenoma; regular MRI surveillance is essential.
4. Additional Medical Therapies
Dopamine Agonists
5. Radiotherapy
Pituitary radiotherapy (stereotactic radiosurgery or fractionated radiotherapy) is reserved for patients with tumour growth or biochemical activity uncontrolled despite surgery and medical therapy. Stereotactic radiosurgery (e.g., Gamma Knife, CyberKnife) is preferred for well-defined residual adenomas <30 mm, not abutting the optic chiasm (≥3–5 mm clearance). Biochemical remission may take 5–15 years to achieve, and hypopituitarism develops in 50–80% of patients over 10 years. Radiotherapy is available at major Australian centres including Peter MacCallum Cancer Centre, Royal North Shore Hospital, and GenesisCare facilities.
Management Algorithm
Monitoring & Surveillance
Lifelong monitoring is mandatory regardless of treatment modality, due to the risk of disease recurrence and comorbidity progression.
| Time Point | Assessment |
|---|---|
| 3 months post-surgery | IGF-1, random GH, OGTT (if IGF-1 elevated), pituitary function panel, MRI pituitary |
| 6 months post-surgery | IGF-1, GH; assess pituitary function; visual fields if macroadenoma |
| 12 months post-surgery | IGF-1, GH, OGTT, pituitary function, MRI; echocardiography if abnormal baseline |
| Annual (years 2–5) | IGF-1, GH; pituitary function if post-surgery; MRI every 1–2 years |
| Annual (lifelong after year 5) | IGF-1; MRI every 2–5 years if stable; ongoing comorbidity monitoring |
| During medical therapy | IGF-1 every 3–6 months; LFTs (pegvisomant: monthly × 6 months, then 6-monthly); gallbladder USS (SRLs annually); glucose monitoring; MRI annually for pegvisomant (tumour surveillance) |
| During radiotherapy | IGF-1 annually; pituitary function panel annually (hypopituitarism risk 50–80% over 10 years); MRI every 1–2 years |
| Colonoscopy | At diagnosis; repeat every 3–5 years (increased colorectal neoplasia risk) |
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Melmed S, Bronstein MD, Chanson P, et al. A Consensus Statement on acromegaly therapeutic outcomes. Nat Rev Endocrinol. 2018;14(9):552–561.
- 2. Katznelson L, Laws ER, Melmed S, et al. Acromegaly: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99(11):3933–3951.
- 3. Giustina A, Barkhoudarian G, Beckers A, et al. Multidisciplinary management of acromegaly: A consensus. Rev Endocr Metab Disord. 2020;21(4):667–678.
- 4. Gadelha M, Kasuki L, Lim DST, Fleseriu M. Systemic complications of acromegaly and the management of patients with persistent disease. Endocr Rev. 2019;40(3):788–825.
- 5. Colao A, Grasso LFS, Giustina A, et al. Acromegaly. Nat Rev Dis Primers. 2019;5(1):20.
- 6. Wass JAH, Turner HE, Adams CBT. The importance of locating a good pituitary surgeon. Pituitary. 2009;12(4):361–364.
- 7. Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum GH and IGF-1 on mortality in acromegaly. Eur J Endocrinol. 2008;159(2):149–155.
- 8. Tritos NA, Biller BMK. Pegvisomant: a growth hormone receptor antagonist used in the treatment of acromegaly. Pituitary. 2017;20(1):129–135.
- 9. Royal Australasian College of Physicians. Clinical practice guidelines for the management of pituitary tumours. Sydney: RACP; 2020.
- 10. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework. Canberra: AIHW; 2023.
- 11. Sherlock M, Ayuk J, Tomlinson JW, et al. Mortality in patients with pituitary disease. Endocr Rev. 2010;31(3):301–342.
- 12. Daly AF, Rixhon M, Adam C, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006;91(12):4769–4775.
- 13. Renehan AG, Bhaskar P, Painter JE, et al. The prevalence and characteristics of colorectal neoplasia in acromegaly. J Clin Endocrinol Metab. 2000;85(9):3417–3424.
- 14. Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021;9(12):847–875.
- 15. Chanson P, Salenave S, Kamenicky P. Acromegaly. In: Handbook of Clinical Neurology. Vol 124. Elsevier; 2014:197–219.