📋 Key Information Summary
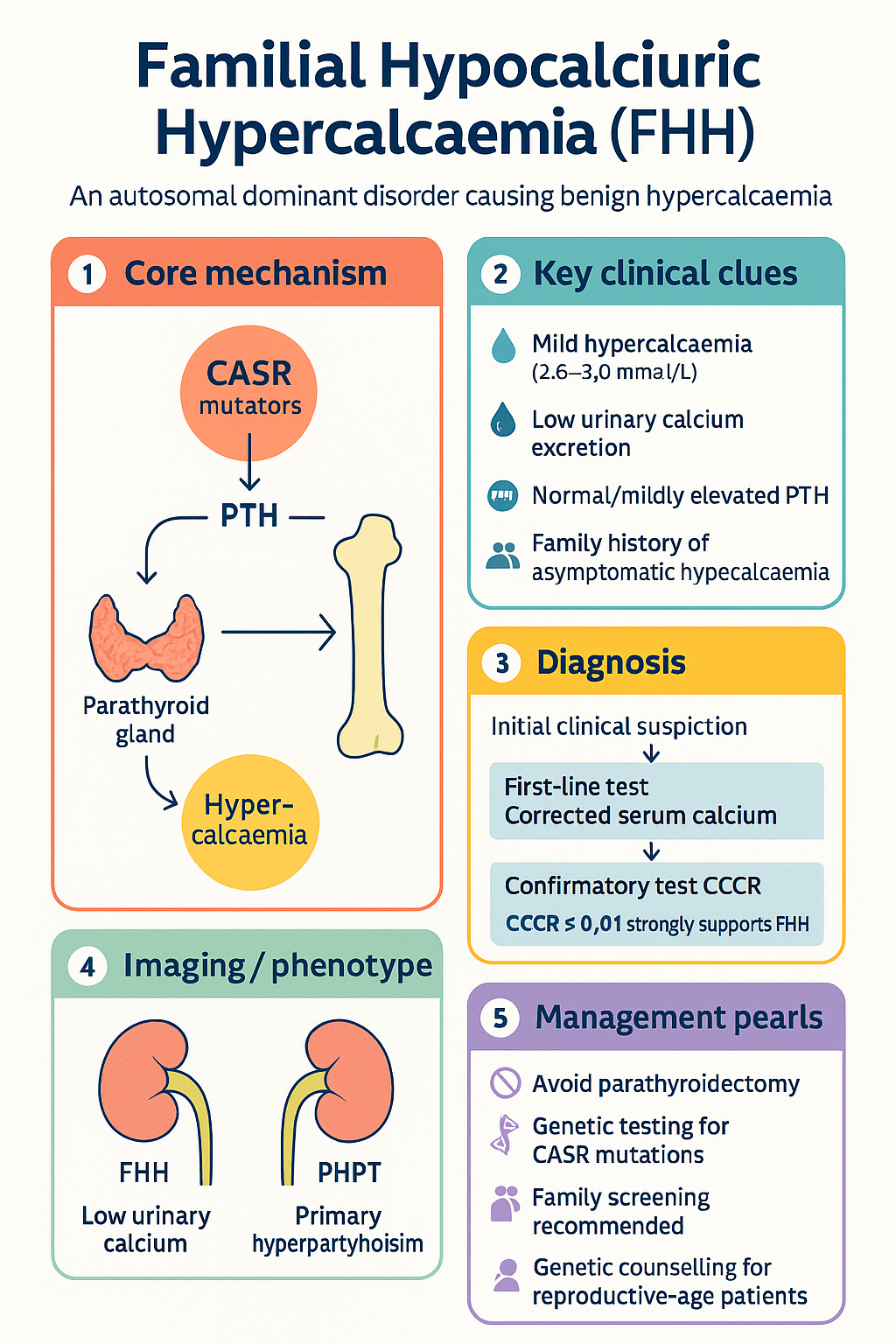
- Familial Hypocalciuric Hypercalcaemia (FHH) is an autosomal dominant disorder caused by inactivating mutations in the calcium-sensing receptor (CASR) gene on chromosome 3q21.1, leading to a raised calcium set-point in the parathyroid glands and kidneys.
- FHH is the most common cause of familial benign hypercalcaemia and is frequently misdiagnosed as primary hyperparathyroidism (PHPT), leading to unnecessary parathyroidectomy.
- The hallmark biochemical feature is low urinary calcium excretion — a calcium/creatinine clearance ratio (CCCR) of <0.01 (or <0.02 depending on assay used) strongly supports FHH over PHPT.
- Patients are typically asymptomatic with mild, lifelong hypercalcaemia (usually corrected calcium 2.6–3.0 mmol/L), normal or mildly elevated PTH, and hypocalciuria.
- Three types are recognised: FHH1 (CASR mutations, ~95% of cases), FHH2 (AP2S1 mutations), and FHH3 (GNA11 mutations).
- Parathyroidectomy is contraindicated in typical FHH — surgery does not cure the hypercalcaemia and exposes patients to unnecessary risk including recurrent laryngeal nerve injury and permanent hypoparathyroidism.
- Genetic testing for CASR mutations is available through Australian molecular genetics laboratories and is recommended when CCCR is equivocal (0.01–0.02) or in paediatric patients.
- No pharmacological treatment is required for the majority of patients. Calcium-lowering therapy is rarely indicated and may cause hypocalcaemia in other family members if applied broadly.
- Family screening with serum calcium, phosphate, PTH, and 24-hour urinary calcium is recommended in all first-degree relatives of confirmed cases.
- FHH Type 3 (FHH3) may present with more pronounced hypercalcaemia and occasional symptomatic disease, requiring closer surveillance.
- All patients of reproductive age should receive genetic counselling given the 50% transmission risk to offspring.
- Differentiation from PHPT is critical: CCCR <0.01, absence of hypercalciuria (<2.5 mmol/24 h in women, <3.0 mmol/24 h in men), and family history of asymptomatic hypercalcaemia favour FHH.
🎧 Audio Brief
Introduction & Australian Epidemiology
Familial Hypocalciuric Hypercalcaemia (FHH) is an autosomal dominant disorder characterised by lifelong, usually asymptomatic mild-to-moderate hypercalcaemia. It is caused by inactivating mutations in genes encoding components of the calcium-sensing receptor (CaSR) signalling pathway, which raises the set-point for calcium-regulated parathyroid hormone (PTH) secretion and renal calcium reabsorption.
FHH is an important diagnostic consideration in any patient presenting with persistent hypercalcaemia accompanied by normal or mildly elevated PTH levels. Its prevalence in the general population is estimated at approximately 1 in 16,000 to 1 in 78,000, though under-recognition is common. Among patients referred for evaluation of hypercalcaemia, FHH accounts for up to 2–5% of cases that might otherwise be labelled as primary hyperparathyroidism.
In Australia, the condition is likely underdiagnosed. Endocrinology referral centres report encountering FHH with increasing frequency as automated serum calcium panels and electronic decision support flag unexpectedly low urinary calcium excretion alongside hypercalcaemia. The true national prevalence is not precisely known due to limited population-based genetic screening data, but extrapolation from European and North American cohorts suggests there may be 250–1,500 affected individuals across Australia, many undiagnosed.
The clinical significance of FHH lies principally in its differential diagnosis from primary hyperparathyroidism (PHPT). Misdiagnosis can lead to inappropriate parathyroidectomy — a procedure that is ineffective in FHH because the fundamental defect is not in the parathyroid glands but in the systemic calcium-sensing mechanism. Avoiding unnecessary surgery is a key quality and safety objective aligned with the Australian Commission on Safety and Quality in Health Care (ACSQHC) Choosing Wisely principles.
Genetics & Pathophysiology
Calcium-Sensing Receptor Biology
The calcium-sensing receptor (CaSR) is a G-protein-coupled receptor (GPCR) expressed predominantly on the surface of parathyroid chief cells and the thick ascending limb of the loop of Henle (TAL) in the kidney. Its primary ligand is extracellular ionised calcium (Ca²⁺). Under normal physiology, rising serum calcium activates CaSR, which:
- In the parathyroid: Suppresses PTH secretion via Gαq-mediated activation of phospholipase C, increased intracellular IP₃, and calcium-dependent inhibition of PTH gene transcription.
- In the kidney: Inhibits paracellular calcium reabsorption in the TAL by reducing the lumen-positive transepithelial voltage (via claudin-14 upregulation and downregulation of the Na⁺-K⁺-2Cl⁻ co-transporter activity), promoting calciuria.
- In bone: Directly stimulates osteoclastic activity and bone resorption at high calcium concentrations (though this is less clinically significant in FHH).
Molecular Genetics of FHH
Three genetically distinct subtypes of FHH are currently recognised, all inherited in an autosomal dominant pattern with high penetrance:
| Subtype | Gene | Chromosome | Protein / Function | Proportion of FHH | Clinical Features |
|---|---|---|---|---|---|
| FHH1 | CASR | 3q21.1 | Calcium-sensing receptor (loss-of-function missense mutations) | ~95% | Mild hypercalcaemia, hypocalciuria; classic benign course |
| FHH2 | AP2S1 | 19q13.3 | Adaptor protein 2 sigma subunit (impairs CaSR endocytosis) | <1% | Similar to FHH1; distinct molecular mechanism |
| FHH3 | GNA11 | 19p13.3 | Gα11 subunit (reduces CaSR signal transduction) | ~3–5% | Higher calcium levels; may occasionally be symptomatic |
Pathophysiological Mechanism
In FHH1, heterozygous loss-of-function mutations in the CASR gene result in a receptor with reduced sensitivity to extracellular calcium. This has two key consequences:
- Parathyroid: The set-point for PTH suppression is shifted to the right, meaning a higher serum calcium concentration is required to suppress PTH. Consequently, PTH is not appropriately suppressed despite hypercalcaemia — it remains normal or mildly elevated, maintaining calcium reabsorption from bone and renal conservation.
- Kidney: Reduced CaSR activation in the TAL impairs the normal calcium-wasting response to hypercalcaemia. Calcium reabsorption is maintained, leading to characteristically low urinary calcium excretion (hypocalciuria). This is the key mechanism generating the diagnostic low CCCR.
The net result is a new, elevated homeostatic set-point for serum calcium: mild hypercalcaemia is maintained lifelong with minimal clinical consequence because the CaSR retains sufficient function to prevent the extreme hypercalcaemia seen in neonatal severe hyperparathyroidism (NSHPT), which occurs with homozygous CASR inactivation.
Distinction from Autosomal Dominant Hypocalcaemia (ADH)
Gain-of-function mutations in CASR cause the mirror-image phenotype — autosomal dominant hypocalcaemia (ADH) — in which the CaSR is over-sensitive to calcium, suppressing PTH at lower-than-normal calcium concentrations and promoting renal calcium wasting. Both FHH and ADH can occur within the same family when different CASR mutations segregate.
Clinical Features & Differentiation from Primary Hyperparathyroidism
Clinical Presentation
The majority of FHH patients are asymptomatic. Hypercalcaemia is typically an incidental finding on routine biochemistry. When the condition is identified, key features include:
- Mild hypercalcaemia (corrected calcium usually 2.60–3.00 mmol/L; reference range 2.10–2.55 mmol/L)
- Normal or mildly elevated intact PTH (within or just above the upper limit of normal)
- Low 24-hour urinary calcium excretion (typically <2.5 mmol/24 h)
- Normal or mildly elevated serum phosphate
- Normal serum magnesium (in most FHH1 patients; mild hypomagnesaemia may occur)
- Normal renal function in most patients
- No nephrolithiasis, nephrocalcinosis, or bone disease attributable to hypercalcaemia
- Family history of asymptomatic hypercalcaemia in ~80% of cases (autosomal dominant inheritance)
Symptoms classically associated with hypercalcaemia — polyuria, polydipsia, constipation, abdominal pain, confusion, nephrolithiasis, and bone pain — are absent in typical FHH. If such symptoms are present, an alternative or concurrent diagnosis (e.g., PHPT, malignancy-associated hypercalcaemia) should be actively sought.
Differentiation from Primary Hyperparathyroidism
Distinguishing FHH from PHPT is the central clinical challenge. The following features help differentiate the two conditions:
| Feature | FHH | Primary Hyperparathyroidism |
|---|---|---|
| Corrected calcium | Mildly elevated (2.60–3.00) | Variable; can be markedly elevated (>2.80) |
| Intact PTH | Normal or mildly elevated (inappropriately normal) | Elevated (inappropriately elevated) |
| 24-h urinary calcium | Low (<2.5 mmol/24 h) | Normal or elevated (>2.5 women, >3.0 men) |
| CCCR | <0.01 | >0.02 (typically 0.02–0.05) |
| Serum phosphate | Normal | Low-normal or low |
| Calcium level over time | Stable lifelong | May progressively rise |
| Symptoms | Asymptomatic | Renal stones, bone loss, fatigue, mood |
| Family history | Multiple relatives with hypercalcaemia | Usually sporadic; familial HPT if MEN1/2A/CDC73 |
| Parathyroid imaging (sestamibi/ultrasound) | Usually negative or non-localising | May localise an adenoma |
| Response to parathyroidectomy | Hypercalcaemia persists or recurs | Curative in >95% of cases |
When to Suspect FHH
Consider FHH in any patient with:
- Asymptomatic mild hypercalcaemia with normal or mildly elevated PTH
- Low 24-hour urinary calcium (<2.5 mmol/24 h) or CCCR <0.01
- A first-degree relative with known hypercalcaemia
- Hypercalcaemia discovered in childhood or adolescence
- Hypercalcaemia persisting after parathyroidectomy
- Failure of hypercalcaemia to resolve after subtotal parathyroidectomy with normal histopathology
Investigations
First-Line Biochemical Assessment
Calcium/Creatinine Clearance Ratio (CCCR) — Detailed Interpretation
The CCCR is the cornerstone test for differentiating FHH from PHPT:
| CCCR Value | Interpretation | Action |
|---|---|---|
| <0.01 | Strongly supportive of FHH | Endocrinology referral; genetic testing if confirmatory; advise against surgery |
| 0.01–0.02 | Equivocal — overlap zone between FHH and PHPT | Repeat 24-h urine collection; CASR genetic testing strongly recommended; endocrinology referral essential |
| >0.02 | Favours PHPT (FHH unlikely but not excluded) | Standard PHPT workup; consider surgery if meeting criteria |
Genetic Testing
Genetic confirmation of FHH is available in Australia through molecular genetics laboratories:
- CASR gene sequencing — the primary test; identifies pathogenic variants in ~65–70% of clinically defined FHH families. Available at major centres including SA Pathology (Adelaide), Royal North Shore Hospital (Sydney), and Victorian Clinical Genetics Services (Melbourne). Medicare rebate may apply under specific genetic testing items (MBS Item 73298 or equivalent).
- AP2S1 and GNA11 sequencing — second-line testing if CASR sequencing is negative. May require referral to a specialist genetics service.
- Genetic testing is recommended in: equivocal CCCR (0.01–0.02), paediatric patients, patients being considered for surgery, and for cascade screening of at-risk family members.
- A negative genetic test does not exclude FHH, as not all pathogenic variants are detectable by current sequencing methods. Clinical and biochemical diagnosis remains valid.
Imaging
Imaging is not routinely indicated in FHH. Specific considerations:
- Sestamibi parathyroid scan / neck ultrasound: Should NOT be performed unless FHH has been excluded, as incidental parathyroid adenomas may be found and inappropriately drive surgical decisions.
- Dual-energy X-ray absorptiometry (DXA): Bone density is typically normal or preserved in FHH. DXA is only indicated if there are additional risk factors for osteoporosis (e.g., postmenopausal status, corticosteroid use).
- Renal ultrasound: Only if there is clinical suspicion of nephrolithiasis or renal impairment; nephrolithiasis is exceedingly rare in FHH.
Family Screening Protocol
Once a proband is identified with confirmed or probable FHH, cascade screening of first-degree relatives is recommended:
- Serum corrected calcium and PTH
- 24-hour urinary calcium and creatinine (for CCCR calculation)
- Serum phosphate and magnesium
- Genetic testing of known familial variant if available (preferred for definitive exclusion in children)
Management
General Principles
FHH is a benign condition in the vast majority of patients. The primary management goal is recognition and avoidance of unnecessary intervention, particularly parathyroidectomy. Management centres on:
- Accurate diagnosis to prevent inappropriate surgery
- Reassurance and education of the patient and family
- Long-term biochemical monitoring (minimal)
- Genetic counselling for affected individuals of reproductive age
- Awareness of the rare symptomatic cases (mainly FHH3)
No Treatment Indicated (Typical FHH)
For the vast majority of patients with FHH1 or FHH2, no pharmacological or surgical treatment is required. Specific points:
- Calcium-lowering agents (e.g., cinacalcet, bisphosphonates) are not routinely indicated.
- There is no dietary calcium restriction recommendation — patients should follow standard Australian dietary guidelines for calcium intake (1,000–1,300 mg/day depending on age and sex).
- Adequate hydration is encouraged but not specifically mandated.
- Thiazide diuretics should be avoided where possible as they may exacerbate hypercalcaemia.
Parathyroidectomy — Not Recommended
Parathyroidectomy is contraindicated in typical FHH. Key points for clinicians:
- Surgery does not correct the underlying calcium-sensing defect and hypercalcaemia persists or recurs post-operatively.
- In a UK series, up to 20% of patients referred for parathyroidectomy for apparent PHPT were subsequently found to have FHH — emphasising the importance of pre-surgical CCCR testing.
- Post-surgical complications include permanent hypoparathyroidism (5–15% risk of bilateral exploration), recurrent laryngeal nerve injury, and the need for lifelong calcium/calcitriol supplementation.
- If a patient has already undergone parathyroidectomy and hypercalcaemia persists, FHH should be investigated and further surgery avoided.
Symptomatic or Marked Hypercalcaemia (Rare — Consider FHH3)
In the rare FHH3 cases or exceptional instances where hypercalcaemia is symptomatic or marked (corrected calcium >3.0 mmol/L), targeted therapies may be considered under specialist endocrinology guidance:
Cinacalcet is the only pharmacological agent with a rational mechanism in FHH (by pharmacologically restoring CaSR sensitivity). However, its use in FHH remains off-label in Australia and should only be considered in exceptional symptomatic cases after specialist review. Case reports and small series demonstrate efficacy in lowering serum calcium in FHH3 patients.
Long-Term Follow-Up
Most patients with confirmed FHH can be managed in primary care with minimal monitoring:
- Annually: Corrected serum calcium and creatinine (integrated into routine GP blood tests).
- Every 3–5 years: Serum PTH if previously borderline elevated.
- As indicated: Serum phosphate, magnesium, 25-hydroxyvitamin D, renal function.
- Endocrinology review if calcium levels rise unexpectedly, symptoms develop, or new features not consistent with FHH emerge (to exclude concurrent PHPT or other pathology).
- Rising calcium levels above the patient's usual stable baseline
- Development of hypercalciuria or nephrolithiasis in a previously stable FHH patient
- Frankly elevated PTH (significantly above the upper limit of normal)
- Localising parathyroid adenoma on imaging
Genetic counselling should be offered to all patients of reproductive age. Given the 50% risk of transmission, pre-conception counselling and cascade genetic testing of children are recommended. There is no evidence that FHH affects fertility or pregnancy outcomes, though calcium levels should be monitored during pregnancy as physiological changes may alter serum calcium.
Concurrent Primary Hyperparathyroidism
Rarely, FHH and PHPT may coexist (estimated prevalence <1:100,000). This is suggested by:
In such cases, surgical intervention may be warranted under expert endocrine surgery guidance, with the understanding that outcomes may be unpredictable.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
While specific prevalence data for FHH in Aboriginal and Torres Strait Islander populations are not available, there are important considerations for equitable diagnosis and management of calcium disorders in these communities.
📚 References
- 1. Hannan FM, Thakker RV. Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Practice & Research Clinical Endocrinology & Metabolism. 2013;27(3):359-371.
- 2. Vargas-Poussou R, et al. CASR mutations causing familial hypocalciuric hypercalcaemia type 1. European Journal of Endocrinology. 2016;175(4):R151-R162.
- 3. Nesbit MA, Hannan FM, Howles SA, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcaemia type 3. Nature Genetics. 2013;45(1):93-97.
- 4. Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G-protein subunit α11 in hypercalcaemia and hypocalcaemia. New England Journal of Medicine. 2013;368(26):2476-2486.
- 5. Christensen SE, Nissen PH, Vestergaard P, Heickendorff L, Brixen K, Mosekilde L. Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow-up study on methods. Clinical Endocrinology. 2008;69(5):713-720.
- 6. Royal Australian College of General Practitioners (RACGP). Red Book: Guidelines for preventive activities in general practice. 9th edn. East Melbourne: RACGP; 2016. [Referenced for family screening principles.]
- 7. The Endocrine Society. The management of asymptomatic primary hyperparathyroidism: an Endocrine Society clinical practice guideline. Journal of Clinical Endocrinology & Metabolism. 2014;99(10):3561-3569.
- 8. Pallais JC, Kemp EH, Bergwitz C, Kannan S, Bilezikian JP. Familial hypocalciuric hypercalcaemia. In: Feingold KR, Anawalt B, Blackman MR, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000–. Updated 2022.
- 9. Australian Commission on Safety and Quality in Health Care (ACSQHC). Australian Atlas of Healthcare Variation. Sydney: ACSQHC; 2017. [Referenced for Choosing Wisely principles regarding unnecessary surgery.]
- 10. Lloyd SE, Pannett AA, Dixon PH, Whyte MP, Bhatt R, Thakker RV. Localization of familial benign hypercalcaemia, Oklahoma variant (FBHOk), to chromosome 19q13. American Journal of Human Genetics. 1999;64(1):189-195.
- 11. NICE Clinical Guideline [CG148]. Hypercalcaemia: diagnosis and management. National Institute for Health and Care Excellence; 2014 (updated 2019).
- 12. Australian Indigenous HealthInfoNet. Overview of Aboriginal and Torres Strait Islander health status 2022. Perth: Australian Indigenous HealthInfoNet, Edith Cowan University; 2023.