📋 Key Information Summary
- Insulinoma is the most common functional pancreatic neuroendocrine tumour, causing endogenous hyperinsulinaemic hypoglycaemia with Whipple's triad.
- Incidence is approximately 1–4 per million per year; most are benign solitary adenomas (>90%) located in the pancreas.
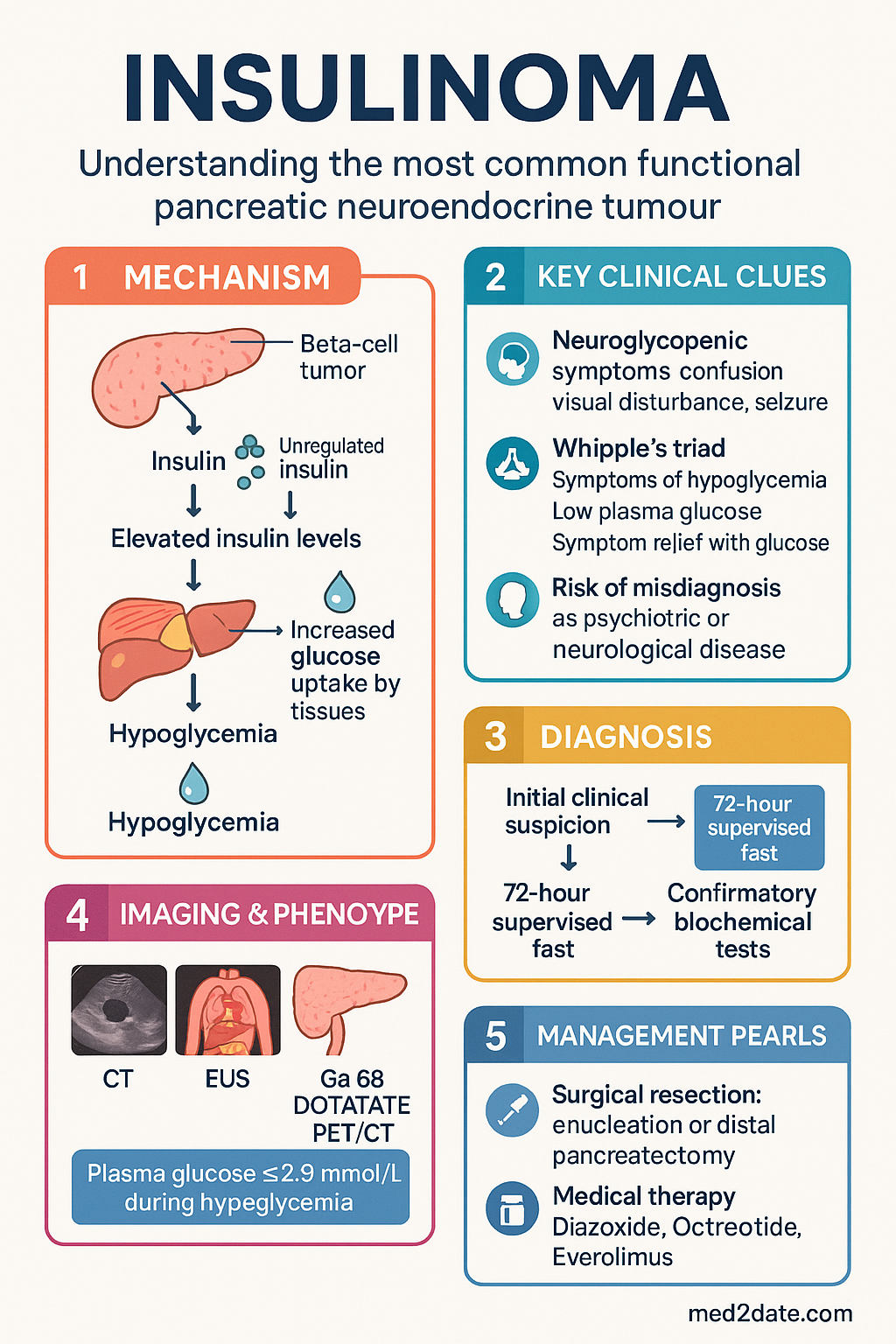
- Whipple's triad — symptoms of hypoglycaemia, documented plasma glucose <2.8 mmol/L, and resolution with glucose administration — must be demonstrated.
- Neuroglycopenic symptoms (confusion, visual disturbance, seizures, loss of consciousness) dominate the clinical picture and may be misdiagnosed as psychiatric or neurological disease.
- The 72-hour supervised fast remains the gold standard provocation test for confirming endogenous hyperinsulinaemic hypoglycaemia.
- Diagnosis requires demonstration of inappropriate insulin, C-peptide, and proinsulin levels at the time of hypoglycaemia, with sulphonylurea screen negative.
- Localisation with CT abdomen, endoscopic ultrasound (EUS), and Ga-68 DOTATATE PET/CT is essential pre-operatively.
- Enucleation or distal pancreatectomy is curative in >90% of benign insulinomas; laparoscopic approach is preferred where feasible.
- Diazoxide (Proglicem®) is the primary medical therapy for inoperable or recurrent insulinoma; octreotide and everolimus are second-line options.
- Malignant insulinomas (<10%) may require debulking surgery, somatostatin analogues, everolimus, peptide receptor radionuclide therapy (PRRT), or systemic chemotherapy.
- Patients with multiple endocrine neoplasia type 1 (MEN1) may have multiple insulinomas requiring subtotal pancreatectomy.
- Aboriginal and Torres Strait Islander patients may face delayed diagnosis due to remote access barriers and limited endocrine specialist availability.
🎧 Audio Brief
Introduction & Australian Epidemiology
Insulinoma is the most common functional pancreatic neuroendocrine tumour (pNET), responsible for endogenous hyperinsulinaemic hypoglycaemia. The condition was first described by William J. Mayo in 1927, and surgical cure has been achievable since the first successful enucleation reported in 1929.
The estimated incidence is 1–4 per million per year, accounting for approximately 1–2% of all pancreatic neoplasms. Insulinomas may occur at any age but peak incidence is in the 4th to 6th decades, with a slight female predominance. In Australia, based on population data, approximately 25–100 new cases are diagnosed annually, though underdiagnosis due to misattribution of neuroglycopenic symptoms to psychiatric or neurological conditions is well recognised.
Over 90% of insulinomas are benign, solitary, small (<2 cm), and intrapancreatic. Malignant insulinomas, defined by the presence of metastases (most commonly hepatic), account for 5–10% of cases. Approximately 4–10% of insulinomas occur in the context of multiple endocrine neoplasia type 1 (MEN1), an autosomal dominant syndrome involving the parathyroids, anterior pituitary, and pancreatic islets.
Early diagnosis and surgical resection remain curative in the vast majority of cases. Delayed recognition leads to recurrent severe hypoglycaemia, falls, seizures, cardiac arrhythmias, and potentially irreversible cognitive impairment.
Pathophysiology & Epidemiology
Pathogenesis
Insulinomas arise from the pancreatic beta cells of the islets of Langerhans. They autonomously secrete insulin in an unregulated fashion, independent of the prevailing blood glucose concentration. This leads to inappropriately elevated circulating insulin levels during fasting states, driving glucose uptake into peripheral tissues and suppressing hepatic gluconeogenesis and glycogenolysis.
The molecular pathogenesis involves:
- Sporadic insulinomas: Somatic mutations in YY1 (Yin Yang 1) at 14q32 are found in approximately 30% of cases. Recurrent mutations in MEN1, DAXX, ATRX, and TSC2 are also described.
- MEN1-associated insulinomas: Germline loss-of-function mutations in the MEN1 tumour suppressor gene (chromosome 11q13) encoding menin, with subsequent somatic loss of heterozygosity. Multiple insulinomas and recurrence are common.
- Malignant transformation: Associated with DAXX/ATRX mutations, alternative lengthening of telomeres (ALT), and higher Ki-67 proliferative indices (>5%).
Anatomical Distribution
Insulinomas are distributed roughly equally throughout the head, body, and tail of the pancreas. Ectopic insulinomas (duodenum, peripancreatic tissue) are exceedingly rare (<1%). Most tumours are <2 cm in diameter at diagnosis, which poses challenges for pre-operative localisation.
Australian Context
In Australia, insulinoma management is concentrated in tertiary centres with multidisciplinary neuroendocrine tumour (NET) teams, including endocrinologists, hepatobiliary surgeons, nuclear medicine physicians, and interventional radiologists. Key centres include Royal North Shore Hospital (Sydney), Austin Health (Melbourne), Royal Adelaide Hospital, and Royal Brisbane and Women's Hospital. The South Australian Cancer Registry and Australian Institute of Health and Welfare (AIHW) data capture pNETs under broader pancreatic cancer coding, making precise incidence data difficult to extract.
Clinical Features
Whipple's Triad
The hallmark diagnostic criterion for insulinoma is the presence of Whipple's triad, first described by Allen Oldfather Whipple in 1938:
- 1. Symptoms consistent with hypoglycaemia (neuroglycopenic and/or adrenergic)
- 2. Concomitant low plasma glucose at the time of symptoms (<2.8 mmol/L; or <3.0 mmol/L in the context of a supervised fast)
- 3. Relief of symptoms upon administration of glucose
Whipple's triad should be demonstrated in a controlled setting (inpatient supervised fast) before proceeding with invasive investigations.
Symptom Classification
Symptoms of insulinoma-related hypoglycaemia can be categorised into two groups:
| Symptom Category | Features | Mechanism |
|---|---|---|
| Neuroglycopenic | Confusion, difficulty concentrating, abnormal behaviour, visual blurring/diplopia, dysarthria, seizures, loss of consciousness, coma | Cerebral glucose deprivation — the brain depends almost exclusively on glucose; cognitive function fails below ~3.0 mmol/L |
| Autonomic (Adrenergic) | Tremor, palpitations, sweating, hunger, anxiety, pallor | Counter-regulatory catecholamine release (adrenaline, noradrenaline) in response to falling glucose |
Temporal Pattern
Symptoms typically occur in the fasting state, particularly in the morning before breakfast or during prolonged physical activity. However, postprandial symptoms can occur. Patients frequently self-medicate with frequent carbohydrate-rich meals, leading to weight gain (reported in 20–40% of patients at diagnosis).
Investigations
Phase 1 — Biochemical Confirmation
The 72-hour supervised fast (also termed the prolonged fast test) remains the gold standard for confirming endogenous hyperinsulinaemic hypoglycaemia.
72-Hour Supervised Fast Protocol
Diagnostic Criteria During Fast
| Parameter | Diagnostic Threshold | Interpretation |
|---|---|---|
| Plasma glucose | <2.8 mmol/L (venous, glucose oxidase) | Confirms hypoglycaemia |
| Serum insulin | ≥36 pmol/L (≥6 μU/mL) at time of hypoglycaemia | Inappropriately non-suppressed |
| C-peptide | ≥200 pmol/L (≥0.6 ng/mL) | Endogenous insulin production confirmed |
| Proinsulin | ≥5 pmol/L | Elevated in insulinoma |
| Beta-hydroxybutyrate | <2.7 mmol/L | Insulin suppresses ketogenesis; low levels confirm hyperinsulinaemia |
| Sulphonylurea screen | Negative | Excludes factitious hypoglycaemia or surreptitious drug use |
Phase 2 — Tumour Localisation
Once biochemical confirmation of hyperinsulinaemic hypoglycaemia is established, tumour localisation is essential for surgical planning. Insulinomas are often small (<2 cm) and require multimodal imaging.
Histopathology
Histological confirmation is obtained from the surgical specimen. Immunohistochemistry is positive for insulin, chromogranin A, and synaptophysin. Ki-67 proliferative index is critical for grading (WHO classification of NETs): G1 (<2%), G2 (2–20%), G3 (>20%).
Management
Surgical Management (Definitive Therapy)
Surgical resection is the definitive treatment for insulinoma and is curative in >90% of benign, sporadic cases. The approach depends on tumour size, location, and number.
| Surgical Approach | Indication | Key Details |
|---|---|---|
| Enucleation | Solitary, superficial tumour <2 cm, not encroaching on main pancreatic duct | Preferred when feasible; lower risk of pancreatic insufficiency. Laparoscopic enucleation increasingly performed in Australia. |
| Distal pancreatectomy ± splenectomy | Tumours in body/tail, or >2 cm, or close to main duct | Spleen-preserving distal pancreatectomy preferred where oncologically appropriate. |
| Pancreaticoduodenectomy (Whipple's) | Large head/uncinate tumours encroaching on bile duct or pancreatic duct | Higher morbidity; performed at high-volume hepatobiliary centres. |
| Subtotal (near-total) pancreatectomy | MEN1-associated multiple insulinomas | Risk of surgical diabetes and exocrine insufficiency; requires lifelong endocrine management. |
Medical Management
Medical therapy is indicated for: (1) pre-operative glycaemic stabilisation, (2) unresectable or metastatic disease, (3) patients unfit for surgery, and (4) bridging while awaiting surgery.
Peptide Receptor Radionuclide Therapy (PRRT)
Lu-177 DOTATATE (Lutathera®) is available in Australia for somatostatin receptor-positive gastroenteropancreatic NETs (GEP-NETs). It may be considered for metastatic insulinoma with high somatostatin receptor expression on Ga-68 DOTATATE PET/CT. Available at specialised centres (Peter MacCallum, Austin Health). TGA-registered; PBS listing requires authority application.
Systemic Chemotherapy
For poorly differentiated (G3) or rapidly progressive malignant insulinoma, platinum-based regimens are considered:
- Cisplatin + Etoposide: Standard for high-grade (G3) neuroendocrine carcinoma
- Temozolomide ± capecitabine: Alternative for well-differentiated progressive disease; activity in pNETs demonstrated in retrospective series
- FOLFOX / FOLFIRI: Reserved for refractory disease; limited evidence
Acute Hypoglycaemia Management
Special Populations
Aboriginal and Torres Strait Islander Health
📚 References
- 1. Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009;94(3):709–728.
- 2. Service FJ, McMahon MM, O'Brien PC, Ballard DJ. Functioning insulinoma — incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. 1991;66(7):711–719.
- 3. Placzkowski KA, Vella A, Thompson GB, et al. Secular trends in the presentation and management of functioning insulinoma at the Mayo Clinic, 1987–2007. J Clin Endocrinol Metab. 2009;94(4):1069–1073.
- 4. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968–977.
- 5. Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513.
- 6. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-DOTATATE for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–135.
- 7. Ramage JK, Ahmed A, Ardill J, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut. 2012;61(1):1–22.
- 8. Australian Institute of Health and Welfare (AIHW). Cancer data in Australia. Canberra: AIHW; 2024. Available from: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia
- 9. Royal Australian College of General Practitioners (RACGP). General practice management of type 2 diabetes: 2016–2018. East Melbourne: RACGP; 2016.
- 10. Jensen RT, Cadiot G, Brandi ML, et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology. 2012;95(2):98–119.
- 11. Abboud B, Boujaoude J. Occult sporadic insulinoma: localization and surgical strategy. World J Gastroenterol. 2008;14(5):657–665.
- 12. Dimitriadis GK, Weickert MO, Randeva HS, et al. Medical management of secretory syndromes. Neuroendocrinology. 2020;110(9–10):813–826.