📋 Key Information Summary
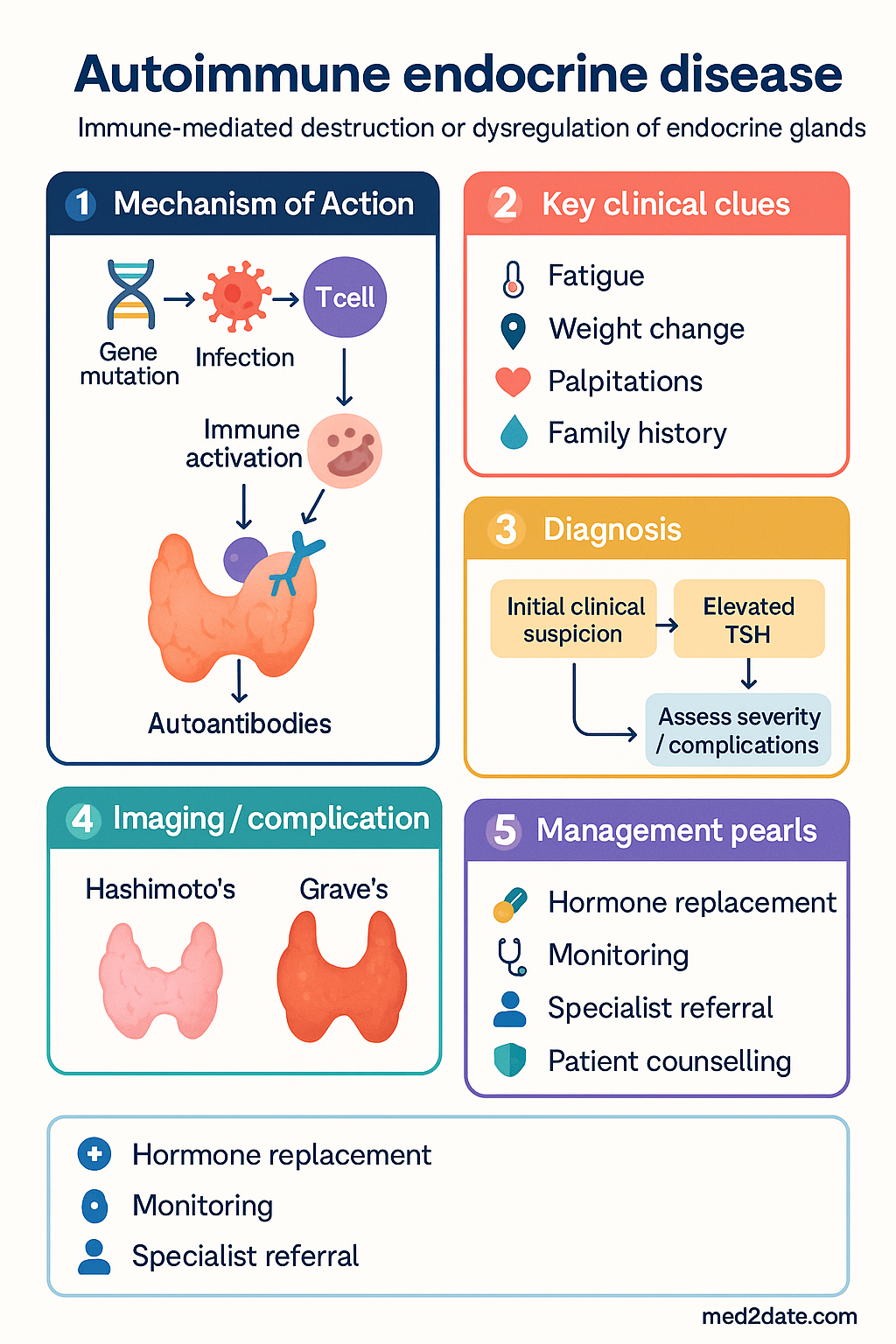
- Autoimmune endocrine diseases result from immune-mediated destruction or dysregulation of endocrine glands and frequently cluster as polyglandular autoimmune syndromes (PGA I and II).
- PGA I presents in childhood with chronic mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency; caused by AIRE gene mutations.
- PGA II (Schmidt syndrome) is the most common form in adults, combining autoimmune adrenalitis with type 1 diabetes and/or autoimmune thyroid disease; HLA-DR3/DR4 associated.
- Key organ-specific autoantibodies include anti-21-hydroxylase (adrenal), anti-GAD65/islet cell (pancreas), anti-TPO/TSH-receptor (thyroid), and anti-tissue transglutaminase (coeliac).
- Screening for associated autoimmune conditions should occur at diagnosis and periodically thereafter — adrenal, thyroid, gonadal, coeliac, and vitamin B₁₂ deficiency.
- Acute adrenal crisis is a medical emergency requiring immediate IV hydrocortisone 100 mg bolus, isotonic saline resuscitation, and identification/treatment of precipitant.
- Autoimmune thyroid disease (Hashimoto's and Graves') is the most prevalent endocrine autoimmunity in Australia, with anti-TPO antibodies present in >90 % of Hashimoto's cases.
- Type 1 diabetes management requires insulin therapy from diagnosis; dual-hormone deficiency (e.g., T1DM + adrenal insufficiency) carries risk of recurrent hypoglycaemia due to cortisol loss.
- Autoimmune hypoparathyroidism (isolated or in PGA I) is treated with calcitriol and calcium supplementation; monitor serum and 24-hour urinary calcium.
- Immunotherapy (steroids, rituximab, mycophenolate) is reserved for select progressive or refractory endocrine autoimmune conditions under specialist guidance.
- Aboriginal and Torres Strait Islander peoples have higher rates of type 1 diabetes and autoimmune thyroid disease; culturally safe screening and GP-led follow-up are essential.
- Pregnancy planning in women with autoimmune endocrine disease requires pre-conception optimisation of thyroid function, adrenal replacement, and glycaemic control (HbA1c <6.5 %).
🎧 Audio Brief
Introduction & Australian Epidemiology
Autoimmune endocrine diseases arise from immune-mediated destruction or aberrant stimulation of endocrine glands. They encompass a spectrum from isolated organ dysfunction (e.g., autoimmune thyroiditis) to multi-gland syndromes classified as polyglandular autoimmune syndromes (PGA). The underlying pathogenesis involves loss of immunological self-tolerance, with autoreactive T cells and organ-specific autoantibodies driving tissue damage.
In Australia, autoimmune endocrine conditions represent a significant disease burden. Autoimmune thyroid disease affects approximately 3–5 % of the adult population, making it one of the most common autoimmune disorders. Type 1 diabetes incidence in Australian children is among the highest globally (~22 per 100 000 per year), with a notable rise over recent decades. Autoimmune adrenal insufficiency (Addison's disease) is rarer (~0.4 per 100 000 per year) but carries life-threatening potential if undiagnosed. The clustering of these conditions — where one autoimmune endocrinopathy predicts another — underscores the importance of longitudinal screening in at-risk populations.
This guideline covers the polyglandular autoimmune syndromes (PGA I and II), the major specific autoimmune endocrinopathies, diagnostic investigations and autoantibody panels, and current Australian management approaches including pharmacotherapy, monitoring strategies, and considerations for special populations.
Polyglandular Autoimmune Syndrome (PGA I & II)
The polyglandular autoimmune syndromes are classified into type I and type II (type III, comprising autoimmune thyroid disease with other non-adrenal organ-specific autoimmunity, is less universally accepted). Distinguishing between PGA I and II is clinically essential, as they differ in genetics, age of onset, and associated conditions.
PGA Type I (APECED)
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is caused by homozygous or compound-heterozygous mutations in the AIRE (autoimmune regulator) gene on chromosome 21q22.3. It follows autosomal recessive inheritance. Onset is typically in childhood (median age 5 years).
| Component | Frequency | Notes |
|---|---|---|
| Chronic mucocutaneous candidiasis (CMC) | >95 % | Usually the first manifestation; oral, skin, nail candidiasis |
| Hypoparathyroidism | 80–90 % | Most common endocrine component |
| Adrenal insufficiency | 60–80 % | Anti-21-hydroxylase antibodies precede clinical disease |
| Gonadal failure | 40–60 % | Primary ovarian or testicular insufficiency |
| Autoimmune hepatitis | 15–20 % | Can be fulminant in children |
| Type 1 diabetes | 10–20 % | Less common than in PGA II |
| Vitiligo / alopecia | 15–30 % | Ectodermal features |
PGA Type II (Schmidt Syndrome)
PGA II is the most common polyglandular syndrome in adults and is associated with HLA-DR3 and HLA-DR4 haplotypes. It comprises autoimmune adrenal insufficiency in combination with at least one of autoimmune thyroid disease (Hashimoto's thyroiditis or Graves' disease) or type 1 diabetes. It has a strong female predominance (F:M ≈ 3:1) and a peak incidence between ages 20 and 40 years.
| Component | Frequency | Notes |
|---|---|---|
| Autoimmune thyroid disease | 70–80 % | Hashimoto's > Graves'; most common component |
| Type 1 diabetes mellitus | 50–60 % | Anti-GAD65 and IA-2 antibodies typical |
| Adrenal insufficiency | Defining component | May develop years after thyroid/diabetes diagnosis |
| Coeliac disease | 2–8 % | Screen with tTG-IgA at diagnosis and periodically |
| Primary ovarian insufficiency | 10–20 % | Anti-ovarian antibodies; FSH elevated |
| Vitiligo | 5–10 % | Anti-melanocyte antibodies |
Specific Autoimmune Endocrinopathies
Autoimmune Adrenal Insufficiency (Addison's Disease)
Autoimmune destruction of the adrenal cortex accounts for 75–90 % of primary adrenal insufficiency in Australia. Anti-21-hydroxylase antibodies are detectable in >85 % of cases. Gradual loss of mineralocorticoid and glucocorticoid function occurs over months to years. By the time clinical adrenal insufficiency is apparent, >90 % of the adrenal cortex has been destroyed.
Key clinical features: Fatigue, weight loss, postural hypotension, hyperpigmentation (ACTH-driven melanocyte stimulation), salt craving, nausea, myalgia. Presentation may be indolent or acute (adrenal crisis).
Autoimmune Thyroid Disease
Hashimoto's thyroiditis (hypothyroid) and Graves' disease (hyperthyroid) represent the two poles of autoimmune thyroid dysfunction. In Australia, autoimmune thyroid disease is the most prevalent endocrine autoimmunity, with a lifetime risk of approximately 5–10 % for women and 1–2 % for men. Hashimoto's is characterised by lymphocytic infiltration and anti-TPO antibodies; Graves' is driven by thyroid-stimulating immunoglobulins (TSI/TRAb).
Type 1 Diabetes Mellitus
Autoimmune destruction of pancreatic beta cells leads to absolute insulin deficiency. Multiple autoantibodies are detectable pre-clinically: anti-GAD65 (most sensitive in adults), anti-IA-2, anti-insulin (IAA, especially in children), anti-ZnT8, and islet cell antibodies (ICA). Presence of ≥2 autoantibodies confers near-100 % lifetime risk of clinical diabetes. Australia has an estimated prevalence of ~140 000 people living with T1DM.
Autoimmune Hypoparathyroidism
Isolated autoimmune hypoparathyroidism is rare outside PGA I. Anti-CaSR (calcium-sensing receptor) antibodies and anti-NALP5 antibodies have been identified. Clinical features include perioral and digital paraesthesia, carpopedal spasm, tetany, seizures, and prolonged QT interval on ECG. Hypocalcaemia with inappropriately low or absent PTH confirms the diagnosis.
Autoimmune Gonadal Failure
Primary ovarian insufficiency (premature ovarian insufficiency, POI) in women and autoimmune orchitis in men may be isolated or part of PGA. In women, anti-ovarian antibodies and anti-steroidogenic enzyme antibodies (anti-17α-hydroxylase, anti-side-chain cleavage enzyme) are associated. Elevated FSH (>25 IU/L) in women <40 years with amenorrhoea confirms the diagnosis. In men, elevated FSH with low testosterone and oligospermia/azoospermia suggests autoimmune testicular failure.
Autoimmune Hypophysitis
Lymphocytic hypophysitis involves T-cell infiltration of the anterior pituitary. It may occur spontaneously (particularly post-partum lymphocytic hypophysitis) or as an immune-related adverse event (irAE) of checkpoint inhibitor therapy (pembrolizumab, nivolumab, ipilimumab). Anti-pituitary antibodies (anti-GH, anti-PIT-1) are detectable in some cases. Imaging shows pituitary enlargement on MRI. Hormonal deficiencies vary; ACTH and TSH loss are most common.
Checkpoint Inhibitor–Related Endocrinopathies
Immune checkpoint inhibitors (ICIs) used in oncology increasingly cause autoimmune endocrinopathies in Australia. Thyroiditis (up to 10 % with anti-PD-1 agents), hypophysitis (up to 10 % with anti-CTLA-4), adrenalitis, and type 1 diabetes are recognised irAEs. These often present acutely and require high clinical suspicion in oncology patients. Management involves hormonal replacement and continuation of immunotherapy when feasible, guided by the treating oncologist.
Investigations & Autoantibodies
Diagnosis of autoimmune endocrine disease relies on a combination of hormonal assays (demonstrating glandular dysfunction) and organ-specific autoantibodies (confirming autoimmune aetiology). The following panels are recommended for screening and diagnosis in the Australian setting.
Recommended Screening Schedule in Known Autoimmune Endocrine Disease
| Screening Test | Frequency | Rationale |
|---|---|---|
| TSH, fT4 | Annually | Autoimmune thyroid disease in any PGA/T1DM patient |
| Morning cortisol (± SST) | If symptoms develop or before planned stress | Adrenal insufficiency may develop late in PGA II |
| tTG-IgA + total IgA | At diagnosis, then every 2–3 years | Coeliac disease screening |
| HbA1c, fasting glucose | Annually (if not T1DM) | Screening for new-onset T1DM in other autoimmune endocrinopathies |
| Vitamin B₁₂, folate | Annually | Autoimmune gastritis / pernicious anaemia association |
| FSH, LH, oestradiol (♀) or testosterone (♂) | If symptoms of hypogonadism | Gonadal failure screening |
Management
Management of autoimmune endocrine disease requires hormone replacement for each deficient gland, treatment of endocrine hyperfunction where present (e.g., Graves' disease), and monitoring for development of additional autoimmune components. Lifelong follow-up is essential.
Adrenal Insufficiency — Glucocorticoid & Mineralocorticoid Replacement
Stress Dosing Protocol
Autoimmune Thyroid Disease — Management
Hypoparathyroidism — Calcium & Vitamin D Replacement
Type 1 Diabetes — Insulin Therapy
All patients with type 1 diabetes require lifelong insulin therapy. Basal-bolus regimens or continuous subcutaneous insulin infusion (CSII / insulin pump) are standard. In the context of PGA II, special attention must be paid to hypoglycaemia risk when coexisting adrenal insufficiency develops — cortisol replacement must be initiated before optimising insulin, as cortisol deficiency impairs gluconeogenesis and hypoglycaemic counter-regulation.
- Basal insulin: insulin glargine (Lantus®, Semglee®) or insulin detemir (Levemir®) — PBS General Benefit.
- Bolus insulin: insulin lispro (Humalog®) or insulin aspart (NovoRapid®) — PBS General Benefit.
- If adrenal insufficiency coexists: ensure hydrocortisone is taken before or at the same time as insulin to avoid post-dose hypoglycaemia.
- Continuous glucose monitoring (CGM) is PBS-subsidised for eligible patients (criteria include T1DM with recurrent severe hypoglycaemia or pregnancy).
Immunomodulatory Therapy
Immunotherapy is not standard for most autoimmune endocrinopathies, as glandular destruction is typically irreversible at diagnosis. However, selected scenarios warrant consideration:
- Early T1DM (within 100 days of diagnosis): Teplizumab (anti-CD3 monoclonal antibody) has shown delay in clinical T1DM onset in high-risk individuals. Currently not PBS-listed in Australia; available through clinical trials and special access schemes.
- Graves' disease refractory to carbimazole/propylthiouracil: Definitive therapy with radioiodine (¹³¹I) or thyroidectomy preferred over immunosuppression.
- Checkpoint inhibitor–related endocrinopathies: High-dose corticosteroids may be used acutely, but permanent hormone replacement is often required. Oncologist guidance essential.
- Rituximab: Case reports in refractory autoimmune hypophysitis and adrenalitis; not standard of care. Specialist use only.
Gonadal Failure — Hormone Replacement
Premature ovarian insufficiency requires oestrogen-progesterone replacement therapy until the age of natural menopause (~50 years). For autoimmune testicular failure, testosterone replacement is indicated. Fertility preservation (oocyte/sperm cryopreservation) should be discussed before irreversible failure.
Monitoring & Long-Term Follow-Up
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Husebye ES, Anderson MS, Kämpe O. Autoimmune polyendocrine syndromes. N Engl J Med. 2018;378(12):1132–1141.
- 2. Betterle C, Garelli S, Presotto F. Diagnosis and classification of autoimmune polyendocrine syndromes (APS). Autoimmun Rev. 2014;13(4–5):455–461.
- 3. Meyer G, Badenhoop K, Linder R. Addison's disease with polyglandular autoimmunity carries a more than 2.5-fold risk for adrenal crises. Eur J Endocrinol. 2016;175(4):317–324.
- 4. Chakera AJ, Vaidya B. Addison disease in adults: diagnosis and management. Am J Med. 2010;123(5):409–413.
- 5. Australian Institute of Health and Welfare (AIHW). Diabetes: Australian facts. Canberra: AIHW; 2023. Cat. no. CVD 86.
- 6. Craig ME, Jefferies C, Dabelea D, Balde N, Seth A, Donaghue KC. ISPAD Clinical Practice Consensus Guidelines 2022: Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatr Diabetes. 2022;23(8):1160–1174.
- 7. Caturegli P, De Remigis A, Rose NR. Hashimoto thyroiditis: clinical and diagnostic criteria. Autoimmun Rev. 2014;13(4–5):391–397.
- 8. Kahaly GJ, Bartalena L, Hegedüs L, Leenhardt L, Poppe K, Pearce SH. 2018 European Thyroid Association guideline for the management of Graves' hyperthyroidism. Eur Thyroid J. 2018;7(4):167–186.
- 9. Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016;101(2):364–389.
- 10. Shun CB, Donaghue KC, Phelan H, Twigg SM, Craig ME. Thyroid autoimmunity in Type 1 diabetes: systematic review and meta-analysis. Diabet Med. 2014;31(2):126–135.
- 11. Australian Government Department of Health. Closing the Gap PBS co-payment measure. Available at: health.gov.au. Accessed 2024.
- 12. Stelmach-Mardas M, Mardas M, Piorunek T, Iber-Kuberacka A. Immune checkpoint inhibitor-related endocrinopathies: a review of the literature. Cancers (Basel). 2023;15(5):1545.
- 13. Herold KC, Bundy BN, Long SA, et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. 2019;381(7):603–613.
- 14. RACGP. Management of type 2 diabetes: a handbook for general practice. Melbourne: RACGP; 2020.
- 15. Australasian Society of Clinical Immunology and Allergy (ASCIA). Autoimmune polyglandular syndromes — clinical update. ASCIA Guidelines 2023.