📋 Key Information Summary
- Phaeochromocytoma is a catecholamine-secreting tumour of the adrenal medulla; paragangliomas are extra-adrenal catecholamine-secreting tumours arising from sympathetic or parasympathetic paraganglia.
- Approximately 10% are bilateral, 10% extra-adrenal, 10% malignant (based on metastatic disease), and 10% hereditary — though recent genetic data suggest hereditary cases may account for up to 30–40%.
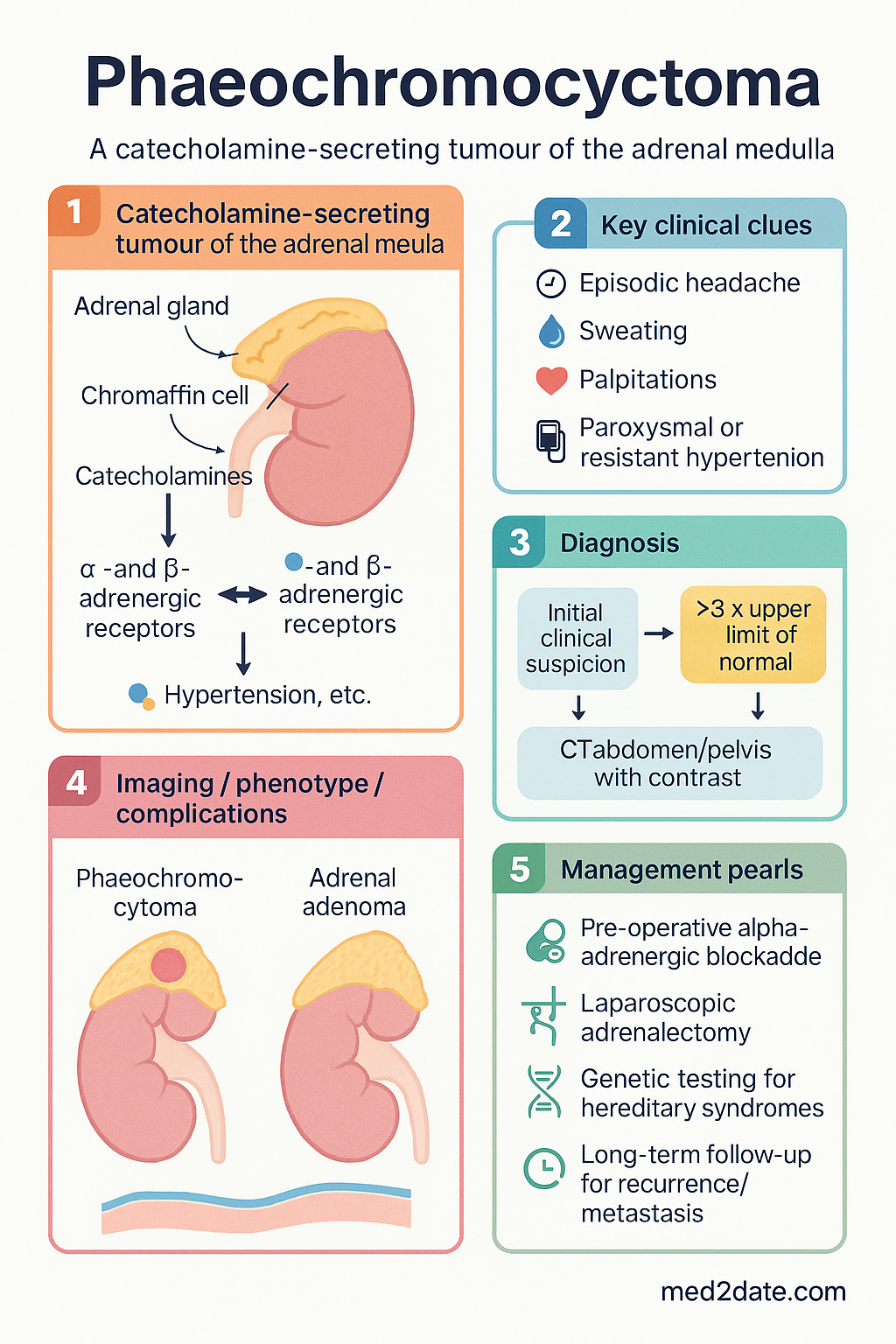
- The classic triad is episodic headache, sweating, and palpitations — but the clinical presentation is heterogeneous; suspicion should be high in paroxysmal or resistant hypertension.
- Plasma free metanephrines are the recommended first-line biochemical test (sensitivity >96%, specificity ~89%); 24-hour urinary fractionated metanephrines and catecholamines are acceptable alternatives.
- CT abdomen/pelvis with contrast is the preferred first-line anatomical imaging; functional imaging with 123I-MIBG or 68Ga-DOTATATE PET/CT is used for metastatic disease, extra-adrenal tumours, or hereditary syndromes.
- Alpha-adrenergic blockade must be commenced at least 10–14 days pre-operatively to prevent intra-operative hypertensive crisis; phenoxybenzamine (non-selective) is the traditional agent; doxazosin (selective α₁) is an alternative.
- Beta-blockade is added only AFTER adequate alpha-blockade to avoid unopposed alpha stimulation and paradoxical hypertensive crisis.
- Pre-operative preparation includes volume expansion (high-salt diet, IV saline), and blood pressure/heart rate targets should be achieved before surgery.
- Laparoscopic adrenalectomy is the standard surgical approach; open surgery is reserved for large (>6 cm) or invasive tumours.
- All patients should undergo genetic testing (minimum panel: RET, VHL, SDHB, SDHD, SDHC, SDHA, MAX, TMEM127, NF1) as hereditary syndromes are present in up to 40% of cases.
- Malignant phaeochromocytoma/paraganglioma (defined by metastatic disease) has a 5-year survival of approximately 50%; cyclophosphamide–vincristine–dacarbazine (CVD) and 131I-MIBG therapy are systemic options.
- Long-term follow-up is mandatory regardless of apparent benign pathology, as recurrence/metastasis can occur decades later — annual biochemical and clinical surveillance is recommended for at least 10 years.
- In Australia, cases should be managed by a multidisciplinary endocrine tumour team including endocrinology, endocrine surgery, anaesthetics, genetics, nuclear medicine, and oncology.
🎧 Audio Brief
Introduction & Australian Epidemiology
Phaeochromocytoma is a catecholamine-secreting neuroendocrine tumour arising from chromaffin cells of the adrenal medulla. When catecholamine-secreting tumours arise from extra-adrenal chromaffin tissue, they are termed paragangliomas. Together, phaeochromocytomas and paragangliomas (PPGL) are considered in the same clinical framework.
The incidence is approximately 2–8 per million population per year, with a peak incidence in the fourth to fifth decades of life. PPGLs are found in approximately 0.1–0.6% of patients with hypertension. In Australia, this equates to roughly 50–100 new diagnoses per year, though improved biochemical testing and imaging have increased detection rates.
The traditional “rule of 10s” (10% bilateral, 10% extra-adrenal, 10% malignant, 10% hereditary) remains a useful heuristic but is an oversimplification. Current data indicate hereditary germline mutations are present in approximately 30–40% of cases, with the proportion even higher in paediatric and bilateral presentations. The term “malignant” is now defined by the presence of metastatic disease (to sites where chromaffin tissue is not normally present) rather than histological features, as no single pathological criterion reliably predicts behaviour.
Undiagnosed phaeochromocytoma carries significant morbidity and mortality due to catecholamine-driven hypertensive crises, arrhythmias, cardiomyopathy, stroke, and multi-organ failure. Prompt biochemical diagnosis, appropriate pre-operative preparation, and surgical resection offer cure in the majority of benign cases.
Pathophysiology & Genetics
Pathophysiology
Phaeochromocytomas arise from chromaffin cells of the adrenal medulla, which are derived from the neural crest. These cells normally synthesise, store, and secrete catecholamines (primarily noradrenaline and adrenaline) under physiological regulation. In phaeochromocytoma, autonomous catecholamine overproduction occurs independent of normal regulatory feedback, leading to sustained or paroxysmal elevation of circulating catecholamines.
The haemodynamic effects of catecholamine excess include:
- Alpha-1 receptor activation → peripheral vasoconstriction → hypertension
- Beta-1 receptor activation → increased heart rate, contractility, and myocardial oxygen demand
- Beta-2 receptor activation → bronchodilation, glycogenolysis, gluconeogenesis
- Metabolic effects: hyperglycaemia, lipolysis, increased basal metabolic rate
Noradrenaline-secreting tumours (predominantly paragangliomas) tend to cause sustained hypertension, whereas adrenaline-secreting tumours (typically adrenal phaeochromocytomas, especially those associated with MEN2) are more commonly associated with paroxysmal hypertension and tachyarrhythmias. Tumour location determines the predominant catecholamine: adrenal phaeochromocytomas contain phenylethanolamine N-methyltransferase (PNMT) and can produce adrenaline, while extra-adrenal paragangliomas generally lack PNMT and secrete noradrenaline.
Genetic Syndromes
At least 20 susceptibility genes have been identified. Genetic testing should be offered to ALL patients with PPGL, as hereditary syndromes are present in 30–40% of cases and have implications for screening, surveillance, and family counselling.
| Syndrome / Gene | Inheritance | PPGL Features | Other Manifestations |
|---|---|---|---|
| VHL (von Hippel–Lindau) | AD | 10–20% develop PPGL; often bilateral; noradrenergic phenotype | Renal cell carcinoma, retinal/cerebellar haemangioblastomas, pancreatic NETs |
| RET (MEN2A/2B) | AD | 50% in MEN2A, 50% in MEN2B; usually bilateral; adrenergic phenotype | Medullary thyroid carcinoma, primary hyperparathyroidism (2A), mucosal neuromas (2B) |
| NF1 (Neurofibromatosis 1) | AD | 1–5% develop PPGL; predominantly adrenal | Neurofibromas, café-au-lait spots, optic gliomas, skeletal abnormalities |
| SDHB | AD | High risk of malignancy (30–50%); often extra-adrenal/paraganglioma | Renal cell carcinoma (type 2), pituitary adenoma |
| SDHD | AD (paternal imprinting) | Often multiple head/neck paragangliomas; low malignancy risk | Head and neck parasympathetic paragangliomas |
| SDHC/SDHA/SDHAF2 | AD | Lower penetrance; head/neck paragangliomas predominance | Variable |
| TMEM127 | AD | Usually benign; predominantly adrenal | Associated with renal cell carcinoma |
| MAX | AD | Often bilateral; moderate malignancy risk | Variable |
| FH | AD | Aggressive; high malignancy risk | Hereditary leiomyomatosis, renal cell carcinoma |
AD = autosomal dominant; NET = neuroendocrine tumour.
In Australia, genetic testing can be performed through public hospital genetics services or private laboratories (e.g., Sonic Genetics, Douglass Hanly Moir). A minimum panel should include RET, VHL, SDHB, SDHD, SDHC, SDHA, MAX, TMEM127, and NF1. Broader next-generation sequencing (NGS) panels encompassing additional genes (FH, MDH2, SLC25A11, DLST, GOT2, IDH3B) are increasingly available.
Clinical Features
The Classic Triad
The classic presentation of phaeochromocytoma comprises three symptoms occurring episodically:
- Headache — typically severe, throbbing, and sudden in onset (present in ~90% of symptomatic episodes)
- Diaphoresis — profuse sweating, often drenching (present in ~60–70%)
- Palpitations — forceful, rapid heartbeat (present in ~50–70%)
When all three are present simultaneously, the positive predictive value for phaeochromocytoma is high. However, the complete triad occurs in only approximately 40% of patients at presentation.
Broader Symptom Profile
Symptoms are often paroxysmal, lasting minutes to hours, and may be triggered by physical exertion, abdominal palpation, micturition (bladder paraganglioma), certain drugs, anaesthesia, or surgery. Between episodes, patients may be asymptomatic.
| Symptom / Sign | Approximate Frequency |
|---|---|
| Headache | 60–90% |
| Diaphoresis | 55–75% |
| Palpitations / tachycardia | 50–70% |
| Hypertension (sustained or paroxysmal) | 80–90% |
| Anxiety / panic / sense of doom | 40–50% |
| Tremor | 30–40% |
| Nausea / vomiting | 25–40% |
| Flushing | 10–30% |
| Chest / abdominal pain | 15–30% |
| Weight loss | 15–30% |
| Pallor | 10–20% |
| Constipation | 10–20% |
| Hyperglycaemia | 10–30% |
Hypertensive Crises
Catecholamine crises may be the initial presentation and can be precipitated by:
- Anaesthesia induction or surgical manipulation
- Bladder instrumentation or micturition (bladder paraganglioma)
- Certain medications: metoclopramide, opioids (morphine), beta-blockers (unopposed alpha), tricyclic antidepressants, contrast dye
- Childbirth
- Vigorous abdominal palpation
Phaeochromocytoma-Induced Cardiomyopathy
Catecholamine excess can cause reversible cardiomyopathy resembling Takotsubo (stress) cardiomyopathy, dilated cardiomyopathy, or catecholamine-induced myocarditis. Recovery typically occurs after tumour resection and alpha-blockade.
Investigations
Biochemical Testing
Biochemical confirmation is essential before any imaging. The Endocrine Society Clinical Practice Guideline (2014, reaffirmed 2023) recommends measurement of plasma free metanephrines or urinary fractionated metanephrines as the first-line test.
Interpretation of Results
Elevation of plasma free normetanephrine to >3× the upper limit of normal has a positive predictive value >95%. Mild elevations (1–3× ULN) require confirmatory testing (repeat measurement, 24-hour urine, or clonidine suppression). Catecholamine levels <ULN effectively exclude phaeochromocytoma.
Anatomical Imaging
Functional Imaging
Functional imaging is indicated when anatomical imaging is negative, for metastatic staging, extra-adrenal disease localisation, or hereditary syndromes with multifocal disease.
Other Investigations
- ECG: May show left ventricular hypertrophy, ST changes, T-wave inversions, prolonged QTc, or arrhythmias
- Echocardiography: Assess for catecholamine cardiomyopathy, especially in patients with heart failure symptoms or troponin elevation
- Troponin: Elevated in catecholamine-induced myocardial injury
- Chromogranin A: Non-specific but may be elevated; useful as a tumour marker for surveillance post-operatively
Risk Stratification & Malignancy Assessment
Malignancy in phaeochromocytoma/paraganglioma is defined solely by the presence of metastases at sites where chromaffin tissue is not normally found (e.g., bone, liver, lung, lymph nodes). No single histological feature reliably predicts malignant behaviour. The PASS (Pheochromocytoma of the Adrenal Scaled Score) and GAPP (Grading System for Adrenal Pheochromocytoma and Paraganglioma) scoring systems provide histological risk estimation but are not diagnostic of malignancy.
Management
Pre-Operative Preparation — Alpha-Blockade
Pre-operative alpha-adrenergic blockade is mandatory and should be initiated at least 10–14 days (optimally 2–4 weeks) before scheduled surgery. The goal is to control hypertension, expand contracted intravascular volume, and prevent intra-operative catecholamine crisis during tumour manipulation.
Beta-Blockade
Beta-blockers are added AFTER 2–3 days of alpha-blockade if persistent tachycardia (>100 bpm) persists.
Volume Expansion
Alpha-blockade causes vasodilation and relative volume contraction. Patients should be encouraged to consume a high-sodium diet (6–8 g salt/day) and liberal oral fluids. IV 0.9% sodium chloride (1–2 L over 12–24 hours) may be administered in the days before surgery, particularly if orthostatic symptoms are significant.
Additional Pre-Operative Agents
Surgical Management
Surgical resection is the definitive treatment for phaeochromocytoma. Laparoscopic adrenalectomy (anterior or lateral transperitoneal, or retroperitoneal) is the standard of care for tumours <6 cm without evidence of local invasion. Open adrenalectomy is indicated for tumours >6 cm, suspected malignancy/invasion, or for bilateral cortical-sparing procedures.
Management of Malignant / Metastatic Disease
For patients with metastatic phaeochromocytoma or paraganglioma, the following systemic and locoregional therapies may be considered:
- 131I-MIBG therapy: High-specific-activity MIBG (Azedra®) has demonstrated efficacy in metastatic disease. Available at select Australian centres via special access schemes.
- 177Lu-DOTATATE PRRT: Peptide receptor radionuclide therapy for somatostatin receptor-positive tumours (e.g., Lutathera®). PBS-listed for gastroenteropancreatic NETs; available off-label for PPGL at specialist centres.
- CVD chemotherapy: Cyclophosphamide 750 mg/m² + Vincristine 1.4 mg/m² + Dacarbazine 600 mg/m² days 1–2 (every 21 days). Response rate ~50% (mostly partial).
- Sunitinib: Tyrosine kinase inhibitor showing activity in metastatic PPGL (37.5 mg daily). Not PBS-listed for this indication.
- Temozolomide: Particularly active in SDHB-mutated tumours (response rate ~30–40%). 200 mg/m² days 1–5 every 28 days.
- Palliative surgery: Cytoreductive surgery for symptom control when feasible.
- External beam radiotherapy: For bone metastases or local symptom control.
Long-Term Surveillance
All patients require lifelong annual biochemical surveillance (plasma free metanephrines) regardless of tumour histology. Minimum recommended follow-up is 10 years, but indefinite surveillance is preferred, as recurrence and metastasis have been reported >20 years post-resection.
| Time Post-Op | Biochemistry | Imaging | Notes |
|---|---|---|---|
| 2–6 weeks | Plasma metanephrines | Nil unless elevated | Confirm biochemical cure |
| Annually (years 1–5) | Plasma metanephrines ± chromogranin A | CT/MRI if hereditary or high-risk | Lifelong for SDHB; annual |
| Annually (years 5–10+) | Plasma metanephrines | Imaging if symptomatic or rising markers | Continue indefinitely for hereditary |
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Lenders JWM, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942.
- 2. Fassnacht M, Assie G, Baudin E, et al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(11):1476–1490.
- 3. Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019;381(6):552–565.
- 4. Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23(34):8812–8818.
- 5. Crona J, Taïeb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr Rev. 2017;38(6):489–515.
- 6. Australian Institute of Health and Welfare (AIHW). Cardiovascular disease, diabetes and chronic kidney disease — Australian facts: Aboriginal and Torres Strait Islander people. Canberra: AIHW; 2015.
- 7. Chen H, Sippel RS, O'Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775–783.
- 8. Hamidi O, Young WF Jr, Iñiguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296–3305.
- 9. Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (Tako-Tsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J. 2008;155(3):408–417.
- 10. Bausch B, Schiavi F, Ni Y, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017;3(9):1204–1212.
- 11. Australian Commission on Safety and Quality in Health Care (ACSQHC). National Safety and Quality Health Service Standards. 2nd ed. Sydney: ACSQHC; 2021.
- 12. Nölting S, Bechmann N, Taïeb DA, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev. 2022;43(2):199–239.