📋 Key Information Summary
- Polycythaemia vera (PV) is a clonal myeloproliferative neoplasm driven by the JAK2 V617F mutation (95%) or, rarely, JAK2 exon 12 mutations, causing constitutive JAK-STAT activation and uncontrolled erythroid, myeloid, and megakaryocyte proliferation.
- Major causes of morbidity and mortality are arterial and venous thrombosis (stroke, MI, portal vein thrombosis, DVT/PE) and transformation to secondary myelofibrosis (post-PV MF) or acute myeloid leukaemia (AML).
- Diagnosis requires WHO 2022 criteria: Hb >165 g/L (males) or >160 g/L (females), or Hct >49% (males) or >48% (females), plus major criterion of BM hypercellularity with panmyelosis OR presence of JAK2 mutation.
- JAK2 V617F mutation testing (peripheral blood) is the single most important diagnostic investigation and is available on MBS.
- First-line therapy is venesection to maintain Hct <45% combined with low-dose aspirin (75–100 mg daily) in all patients unless contraindicated.
- Cytoreduction with hydroxycarbamide (hydroxyurea, Hydrea®) is indicated for high-risk patients (age >60 or prior thrombosis) and selected intermediate-risk patients.
- Ruxolitinib (Jakavi®) is second-line cytoreductive therapy for hydroxycarbamide intolerance/resistance and is PBS Authority Required.
- Risk stratification drives treatment intensity: low-risk patients receive phlebotomy + aspirin only; high-risk patients require cytoreduction.
- Key safety alert: uncontrolled PV (Hct >52%) dramatically increases thrombotic risk; every effort must be made to maintain Hct <45%.
- Aboriginal and Torres Strait Islander peoples may have higher burden of cardiovascular risk factors compounding PV-related thrombosis risk; culturally safe, community-based follow-up is essential.
- Monitor for disease transformation — worsening constitutional symptoms, increasing splenomegaly, or new cytopenias should prompt reassessment for post-PV myelofibrosis or AML.
- Pregnancy requires specialist haematology input; venesection and low-dose aspirin are generally continued; interferon-alpha is the preferred cytoreductive agent if required.
Introduction & Australian Epidemiology
Polycythaemia vera (PV) is a chronic myeloproliferative neoplasm (MPN) characterised by clonal proliferation of haematopoietic precursors, leading to erythrocytosis with or without leucocytosis and thrombocytosis. It is one of three classical Philadelphia-chromosome-negative MPNs, alongside essential thrombocythaemia (ET) and primary myelofibrosis (PMF).
In Australia, PV accounts for approximately 2–3 new diagnoses per 100,000 population per year. The median age at diagnosis is 60–65 years, with a slight male predominance (M:F ratio approximately 1.2:1). The disease is rare under 40 years of age, though JAK2 exon 12-mutated PV can present earlier. Australian registry data from Cancer Council Victoria and the AIHW confirm that PV incidence is rising in parallel with increased use of routine full blood count (FBC) screening and JAK2 mutation testing in general practice.
The principal clinical concern in PV is thrombotic complication, which accounts for approximately 40–50% of deaths. In the landmark CYTO-PV trial, patients randomised to a haematocrit (Hct) target of <45% had significantly fewer major thrombotic events and cardiovascular deaths compared with those maintained at 45–50%. This finding underpins the current therapeutic standard of maintaining Hct <45% in all patients with PV.
With appropriate treatment, median survival in PV exceeds 15–20 years, though this is still reduced compared with the age-matched general population. Transformation to secondary myelofibrosis occurs in approximately 10–15% of patients over 10–15 years, and progression to AML in 2–5%.
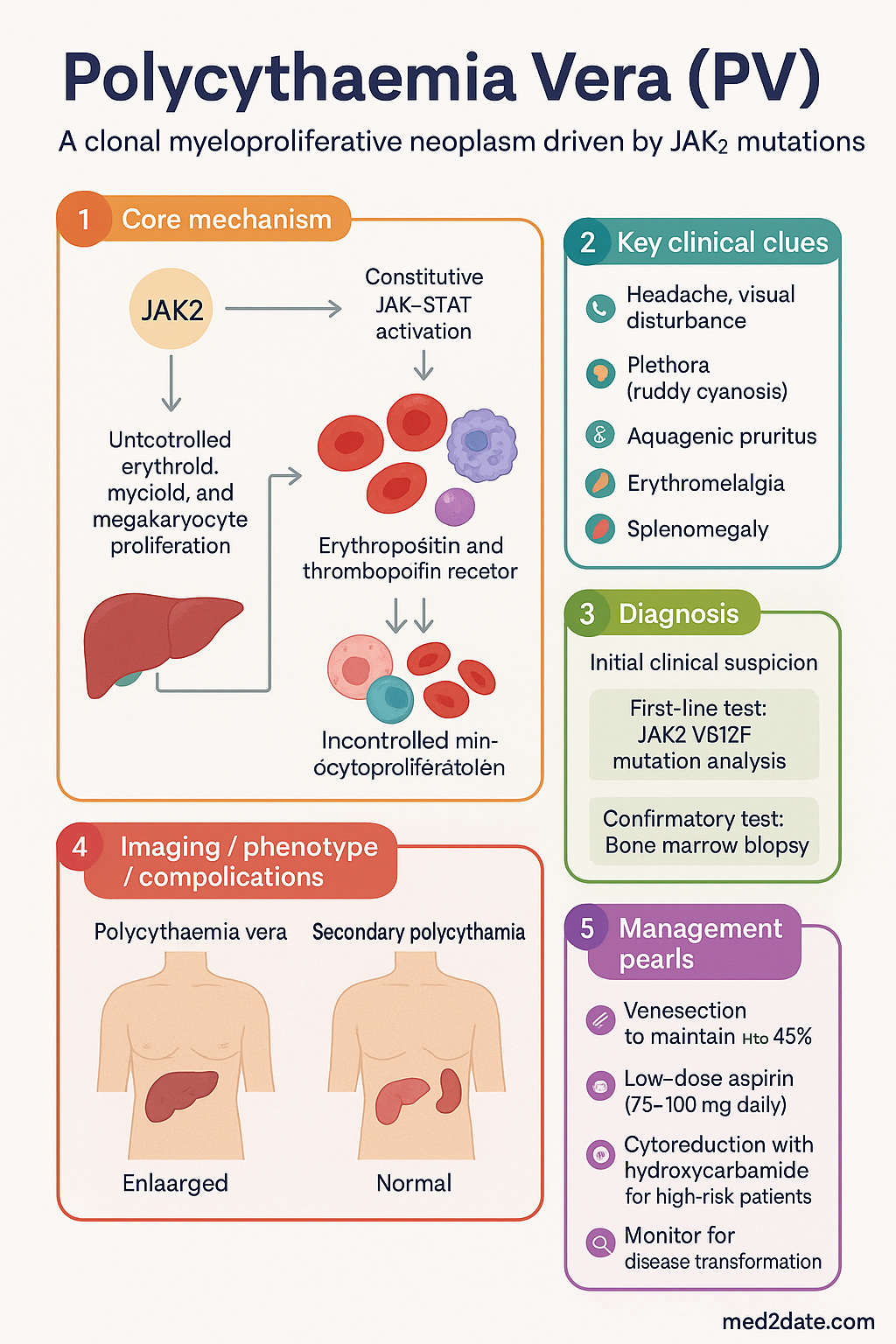
Pathogenesis — JAK2 V617F Mutation
The pathogenesis of PV is driven by somatic mutations in the Janus kinase 2 (JAK2) gene, which encodes a cytoplasmic tyrosine kinase essential for haematopoietic growth factor receptor signalling. Two mutation types are recognised:
Downstream Consequences of JAK2 Activation
- Erythropoietin-independent erythropoiesis: Mutant JAK2 mimics constitutive EPO receptor signalling, causing erythroid progenitor proliferation independent of erythropoietin levels. Serum EPO is characteristically low.
- Thrombopoietin-independent thrombopoiesis: Megakaryocyte proliferation is driven by constitutive TPO receptor signalling.
- Granulocyte proliferation: Myeloid expansion contributes to leucocytosis in many patients.
- Increased neutrophil-platelet interaction: Activated leucocytes and platelets promote a prothrombotic milieu, explaining the disproportionate thrombosis risk relative to Hct alone.
- Low serum EPO: A key distinguishing feature from secondary (reactive) polycythaemia, where EPO is normal or elevated.
Additional Somatic Mutations
Co-mutations in epigenetic regulators (TET2, ASXL1, DNMT3A, EZH2) and spliceosome genes (SRSF2) are found in 20–40% of PV patients and may influence disease phenotype, thrombosis risk, and transformation risk. Mutations in ASXL1, SRSF2, IDH1/2, and RUNX1 are associated with inferior survival and higher rates of leukaemic transformation.
Clinical Features & WHO 2022 Diagnostic Criteria
Clinical Presentation
Approximately 30–40% of PV patients are asymptomatic at diagnosis, detected incidentally on FBC. Symptomatic patients may present with:
- Headache, visual disturbance (scintillating scotomata), dizziness, tinnitus: due to hyperviscosity
- Plethora (ruddy cyanosis): facial and conjunctival erythema — a classic but not universal sign
- Aquagenic pruritus: intense itching after warm bathing or showering, occurs in 40–65% of patients; pathognomonic when present
- Erythromelalgia: burning pain and erythema of the extremities (typically hands/feet); caused by platelet-mediated arteriolar microthrombi
- Splenomegaly: present in 40–70% at diagnosis; hepatomegaly in 30–40%
- Fatigue, night sweats, weight loss: constitutional symptoms present in ~30%
- Thrombotic events: stroke, TIA, MI, peripheral arterial thrombosis, portal vein thrombosis (Budd-Chiari syndrome), mesenteric vein thrombosis, DVT, PE
- Haemorrhagic events: paradoxically, bleeding (gum bleeding, epistaxis, GI haemorrhage) may occur due to acquired von Willebrand syndrome at very high platelet counts (>1000 × 10⁹/L)
- Gout: secondary hyperuricaemia from increased cell turnover
WHO 2022 Diagnostic Criteria for PV
The diagnosis of PV requires meeting all three major criteria, or the first two major criteria plus the minor criterion.
| Criterion | Requirement |
|---|---|
| Major criterion 1 — Haemoglobin/Hct threshold | Hb >165 g/L (males) or >160 g/L (females) OR Hct >49% (males) or >48% (females) |
| Major criterion 2 — Bone marrow biopsy | Hypercellularity for age with panmyelosis (erythroid, granulocytic, and megakaryocytic proliferation). Grading of reticulin fibrosis (grade 0–1 recommended at diagnosis). |
| Major criterion 3 — JAK2 mutation | Presence of JAK2 V617F mutation or JAK2 exon 12 mutation |
| Minor criterion | Subnormal serum erythropoietin level |
Investigations
Investigation follows a structured approach: confirm erythrocytosis, exclude reactive causes, establish clonality, and risk-stratify.
Laboratory Investigations
Excluding Reactive (Secondary) Polycythaemia
Imaging
- Abdominal ultrasound: Assess spleen size (normal <13 cm craniocaudal length). Document hepatomegaly. MBS item 55043.
- Cross-sectional imaging (CT/MRI): If ultrasound findings equivocal or to assess for thrombotic complications (portal vein, hepatic veins, mesenteric veins).
Risk Stratification
Risk stratification in PV determines treatment intensity. The conventional two-tier model classifies patients as low-risk or high-risk based on age and thrombosis history. More recent models incorporate additional cardiovascular risk factors.
Management
Management of PV has three pillars: (1) reduction of Hct to <45% via venesection, (2) antiplatelet therapy with low-dose aspirin, and (3) cytoreductive therapy for high-risk and selected intermediate-risk patients.
Pillar 1 — Venesection (Phlebotomy)
Venesection is the cornerstone of PV therapy and is indicated in all patients. The target Hct is <45%, based on the CYTO-PV trial (Marchioli et al., NEJM 2013).
- Initial phase: Venesection of 450 mL (or 250 mL for patients <50 kg) every 1–2 weeks until Hct <45%.
- Maintenance phase: Venesection every 2–3 months to maintain Hct <45%. Frequency guided by FBC monitoring.
- Iron depletion: Repeated venesection inevitably leads to iron deficiency. This is expected and generally well tolerated. Do not supplement iron unless clinically symptomatic.
- Logistics: Venesection can be performed in hospital day units, community pathology centres, or via Australian Red Cross Lifeblood (where eligible). MBS item 13700 (therapeutic venesection).
Pillar 2 — Low-Dose Aspirin
Low-dose aspirin (75–100 mg daily) is recommended for all patients with PV unless contraindicated, based on the ECLAP trial (Landolfi et al., NEJM 2004). Aspirin reduces the risk of arterial and venous thrombotic events by approximately 60%.
- Dose: 75–100 mg orally once daily (e.g., Aspirin 100 mg, Cartia®).
- Contraindications: Active bleeding, history of GI haemorrhage (unless on PPI), severe uncontrolled hypertension, platelet count >1500 × 10⁹/L (risk of acquired vWD bleeding), aspirin allergy.
- Acquired von Willebrand syndrome: At very high platelet counts (>1000 × 10⁹/L), acquired vWD may develop. Consider checking vWF antigen/activity before commencing aspirin if platelets >1000 × 10⁹/L.
- PBS status: General Benefit (over-the-counter in Australia).
Pillar 3 — Cytoreductive Therapy
Cytoreduction is indicated for high-risk patients (age ≥60 or prior thrombosis) and may be considered for intermediate-risk patients with inadequate disease control on venesection alone.
Symptomatic Management
| Symptom | Management |
|---|---|
| Aquagenic pruritus | Cetirizine 10 mg OD; paroxetine 20 mg OD (off-label); narrowband UVB phototherapy; aspirin may help; ruxolitinib is particularly effective |
| Erythromelalgia | Low-dose aspirin is highly effective (often resolves within hours); also control Hct and platelet count |
| Gout / hyperuricaemia | Allopurinol 100–300 mg OD; colchicine 0.5 mg for acute flares (renal adjustment required) |
| Fatigue | Optimise Hct; rule out iron deficiency; graded exercise programme; consider ruxolitinib if refractory |
| Splenomegaly discomfort | Cytoreduction; avoid contact sports; ruxolitinib for refractory splenomegaly |
Monitoring
Lifelong monitoring is essential in PV. The frequency and intensity of monitoring depends on disease phase, risk category, and treatment.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
While PV is not known to be more prevalent in Aboriginal and Torres Strait Islander peoples, the management of PV is significantly influenced by the higher background prevalence of cardiovascular risk factors, limited specialist access in remote communities, and culturally specific health considerations.
📚 References
- 1. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. doi:10.1056/NEJMoa1208500
- 2. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–124. doi:10.1056/NEJMoa035572
- 3. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. doi:10.1056/NEJMoa1409002
- 4. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 2017;18(1):88–99. doi:10.1016/S1470-2045(16)30558-7
- 5. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703–1719. doi:10.1038/s41375-022-01613-1
- 6. Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. 2024;99(3):475–494. doi:10.1002/ajh.27189
- 7. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015;5(11):e369. doi:10.1038/bcj.2015.94
- 8. Australian Institute of Health and Welfare (AIHW). Cardiovascular disease in Aboriginal and Torres Strait Islander people. Cat. no. CVD 80. Canberra: AIHW; 2023.
- 9. Geyer HL, Scherber RM, Kosiorek H, et al. Symptomatic profiles of patients with polycythemia vera: implications of inadequately controlled disease. J Clin Oncol. 2016;34(2):151–159. doi:10.1200/JCO.2015.62.9337
- 10. McMullin MF, Harrison CN, Ali S, et al. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology guideline. Br J Haematol. 2019;184(2):176–191. doi:10.1111/bjh.15648
- 11. Grunwald MR, Burke JM, Kuter DJ, et al. Real-world risk assessment and treatment patterns in patients with polycythemia vera. Ann Hematol. 2022;101(3):581–590. doi:10.1007/s00277-021-04727-x
- 12. Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002;100(13):4272–4290. doi:10.1182/blood-2001-12-0349