📋 Key Information Summary
- Microangiopathic haemolytic anaemia (MAHA) is a clinicopathological syndrome defined by intravascular red cell fragmentation producing schistocytes on peripheral blood film, combined with thrombocytopenia and elevated lactate dehydrogenase (LDH).
- The underlying mechanism is mechanical shearing of erythrocytes as they traverse microvessels occluded by fibrin strands, platelet thrombi, or endothelial injury.
- The five major causes are thrombotic thrombocytopenic purpura (TTP), haemolytic uraemic syndrome (HUS), disseminated intravascular coagulation (DIC), malignant hypertension, and drug-/transplant-related microangiopathy.
- TTP is a medical emergency — plasma exchange must be initiated within 4–8 hours of clinical suspicion; mortality without treatment exceeds 90%.
- The PLASMIC score helps stratify pre-test probability of TTP: a score ≥5 confers high risk (positive predictive value >90%) and should trigger immediate plasma exchange before ADAMTS13 result.
- Severe ADAMTS13 deficiency (<10% activity) is diagnostic of TTP; ADAMTS13 turnaround in Australian centres is typically 24–72 hours.
- Complement-mediated HUS (atypical HUS) is treated with eculizumab (Soliris®), now PBS-listed in Australia as an Authority Required medication.
- DIC-related MAHA mandates treatment of the underlying trigger (sepsis, trauma, obstetric complication) with supportive platelet and cryoprecipitate transfusions guided by serial coagulation assays.
- Blood film review is the cornerstone diagnostic test — the presence of ≥2 schistocytes per high-power field in the context of thrombocytopenia and haemolysis is highly suggestive of MAHA.
- Aboriginal and Torres Strait Islander peoples experience a disproportionate burden of sepsis-related DIC and hypertensive emergencies; early recognition and culturally safe care pathways are essential.
- All cases of suspected MAHA should have urgent FBC, blood film, LDH, haptoglobin, coagulation studies, ADAMTS13, renal function, and a peripheral blood film reviewed by a haematologist.
- Rituximab (Mabthera®) is used as adjunctive therapy in refractory or relapsing TTP and is PBS-listed for this indication under the Immunoglobulin-Resistant TTP protocol.
Introduction & Australian Epidemiology
Microangiopathic haemolytic anaemia (MAHA) is a life-threatening haematological syndrome in which red blood cells are mechanically fragmented as they pass through partially occluded arterioles and capillaries. The hallmark is the identification of schistocytes — irregularly shaped red cell fragments — on the peripheral blood film, accompanied by evidence of haemolysis (elevated LDH, low haptoglobin, elevated unconjugated bilirubin) and consumptive thrombocytopenia. The reticulocyte count is typically elevated, reflecting compensatory erythropoiesis.
MAHA is not a diagnosis in itself but rather a pathological finding that demands urgent identification of the underlying aetiology. The principal causes encountered in Australian practice include thrombotic thrombocytopenic purpura (TTP), typical and atypical haemolytic uraemic syndrome (HUS), disseminated intravascular coagulation (DIC), malignant hypertension, and post-transplant or drug-associated thrombotic microangiopathy (TMA).
In Australia, the estimated incidence of TTP is approximately 3–6 cases per million population per year, with a peak in middle-aged women. The Australasian Society of Blood Transfusion (ASBT) and National Blood Authority data indicate that approximately 150–200 plasma exchange procedures are performed annually for TTP across major metropolitan centres. HUS — particularly Shiga toxin-producing E. coli (STEC) HUS — peaks in summer months and disproportionately affects young children, with sporadic outbreaks reported from childcare centres and undercooked meat exposure.
DIC-related MAHA is the most common cause encountered in Australian tertiary intensive care units, complicating severe sepsis (particularly meningococcaemia and Clostridium perfringens bacteraemia), major trauma, and obstetric catastrophes such as placental abruption and amniotic fluid embolism. Aboriginal and Torres Strait Islander peoples experience a two- to four-fold higher incidence of sepsis-related DIC, driven by higher rates of community-acquired infections, delayed healthcare access, and comorbid chronic disease.
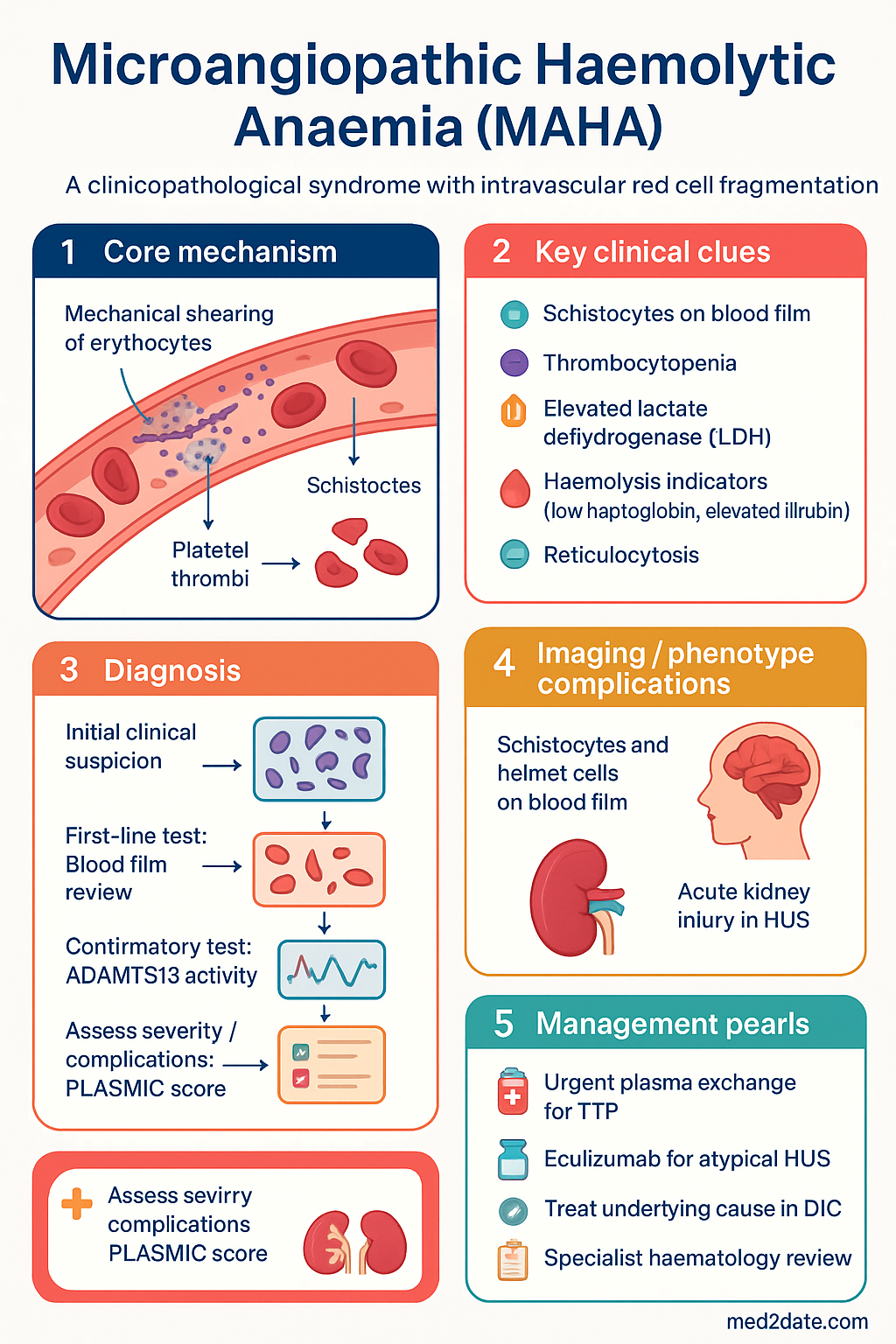
Pathogenesis & Causes
The common pathogenic endpoint in all forms of MAHA is mechanical shearing of erythrocytes within the microvasculature. This occurs when fibrin strands, platelet microthrombi, or damaged endothelium create a partial physical barrier across which red cells are forced at high shear stress, producing the characteristic fragmented morphology. The process is intravascular and leads to haemoglobinuria when severe.
Major Causal Categories
| Category | Mechanism | Key Features |
|---|---|---|
| Thrombotic thrombocytopenic purpura (TTP) | Severe ADAMTS13 deficiency (<10%) → accumulation of ultra-large von Willebrand factor (vWF) multimers → platelet-rich thrombi in arterioles | Pentad (fever, MAHA, thrombocytopenia, renal impairment, neurological symptoms); neurological features predominate; renal involvement typically mild |
| Typical HUS (STEC-HUS) | Shiga toxin (Stx1/Stx2) → endothelial injury → complement activation → fibrin deposition in glomerular capillaries | Prodromal bloody diarrhoea; children <5 years; severe acute kidney injury; often O157:H7 or O104:H4 |
| Atypical HUS (aHUS) | Dysregulated complement alternative pathway (mutations in CFH, CFI, MCP, C3, CFHR1/3, thrombomodulin) | No diarrhoeal prodrome; relapsing–remitting course; severe AKI; complement-mediated |
| Disseminated intravascular coagulation (DIC) | Systemic activation of coagulation → widespread fibrin deposition → secondary fibrinolysis | Consumptive coagulopathy (↑PT, ↑aPTT, ↓fibrinogen, ↑D-dimer); bleeding and thrombosis simultaneously; underlying trigger (sepsis, trauma, obstetric) |
| Malignant hypertension | Endothelial injury from shear stress in arterioles → fibrinoid necrosis | BP typically >180/120 mmHg with end-organ damage; papilloedema; acute kidney injury; encephalopathy |
| Drug-induced TMA | Drug-dependent anti-ADAMTS13 antibodies (e.g. quinine, cyclosporin, tacrolimus) or direct endothelial toxicity (gemcitabine, bevacizumab, VEGF inhibitors) | Temporal relationship to drug exposure; ADAMTS13 may or may not be severely reduced |
| Post-transplant TMA | Calcineurin inhibitor toxicity + complement activation post-HSCT or solid organ transplant | 10–25% post-HSCT; worse prognosis in allo-HSCT; consider complement-mediated component |
| Pregnancy-associated TMA | Pre-eclampsia/eclampsia, HELLP syndrome, TTP in pregnancy, aHUS unmasked postpartum | Onset usually 2nd–3rd trimester or early postpartum; maternal and foetal mortality risk |
Blood Film — Schistocytes, Helmet Cells & Fragmented Erythrocytes
The peripheral blood film is the single most important bedside investigation in suspected MAHA. A trained haematologist or haematology scientist should review the film, as automated analysers may undercount or misclassify schistocytes.
Morphological Features
- Schistocytes: Irregularly shaped red cell fragments with angular borders, often described as "helmet cells," triangular fragments, or crescent-shaped cells. They lack central pallor and vary in size (microspherocytes to large fragments).
- Helmet cells: A subtype of schistocytes resembling the outline of a medieval helmet; formed when a portion of the red cell is sheared off against fibrin strands.
- Keratocytes (bite cells): Red cells with one or more semi-circular bites removed; may be confused with schistocytes but are more commonly associated with oxidative haemolysis (G6PD deficiency, Heinz body haemolytic anaemia).
- Microspherocytes: Small, dense spherocytes without central pallor; generated by partial fragmentation and membrane loss.
Quantitative Thresholds
| Schistocyte Count | Interpretation |
|---|---|
| <1% of red cells | Normal or non-specific; may be seen post-splenectomy, in severe burns, or as a laboratory artefact |
| 1–2% (≥2 per HPF) | Suspicious for MAHA in the clinical context of thrombocytopenia and haemolysis — warrants urgent investigation |
| >2–5% | Highly suggestive of TMA; correlate with ADAMTS13, DIC panel, renal function |
| >5% | Severe MAHA; near-diagnostic in the setting of thrombocytopenia; immediate plasma exchange should be considered pending ADAMTS13 |
Associated Blood Film Findings
- Thrombocytopenia (platelet count typically <100 × 10⁹/L, often <30 × 10⁹/L in TTP)
- Polychromasia (reflecting reticulocytosis)
- Nucleated red blood cells (nRBCs) — seen in severe haemolysis
- Leucoerythroblastic picture — concerning for marrow infiltration or severe sepsis
- Neutrophilia with toxic granulation — suggests sepsis/DIC aetiology
Investigations & Diagnosis
The diagnostic workup of MAHA must proceed rapidly and in parallel with empirical management. Key objectives are: (1) confirm haemolytic aetiology with fragmentation, (2) exclude common mimics (autoimmune haemolytic anaemia, PNH), (3) identify the specific TMA subtype, and (4) stratify urgency.
First-Line Investigations (Perform Immediately)
Second-Line / Guided Investigations
Risk Stratification & Severity Scoring
PLASMIC Score for Pre-Test TTP Probability
The PLASMIC score is the most validated tool for estimating the likelihood of severe ADAMTS13 deficiency in patients presenting with MAHA and thrombocytopenia. It can be calculated before ADAMTS13 results are available.
| Criterion | Points |
|---|---|
| Platelet count <30 × 10⁹/L | 1 |
| Haemolysis (reticulocyte count >2.5%, haptoglobin undetectable, or indirect bilirubin >2 mg/dL) | 1 |
| Active malignancy | 0 (no = 1) |
| History of solid-organ or stem cell transplant | 0 (no = 1) |
| No history of recent surgery | 1 |
| No bleeding within past 1 month | 1 |
| INR <1.5 | 1 |
| MCHC ≥340 g/L | 1 |
| MCV ≥90 fL | 1 |
| Creatinine <1.5 mg/dL (<133 µmol/L) | 1 |
| ANA titre <1:160 | 1 |
ISTH DIC Score
For suspected DIC-related MAHA, the International Society on Thrombosis and Haemostasis (ISTH) scoring system provides a validated approach:
- Platelet count: >100 = 0; <100 = 1; <50 = 2
- Elevated fibrin marker (D-dimer/FDP): no increase = 0; moderate = 2; strong = 3
- Prolonged PT: <3 sec = 0; 3–6 sec = 1; >6 sec = 2
- Fibrinogen: >1.0 g/L = 0; <1.0 g/L = 1
- Score ≥5: compatible with overt DIC; <5: suggestive but not overt — repeat in 6–8 hours
Management — Treat the Underlying Cause
The management of MAHA is entirely dependent on identifying and treating the underlying aetiology. There is no single therapy for MAHA itself — treatment of the driving pathology (whether it is TTP, HUS, DIC, or another cause) will resolve the microangiopathic process.
1. Thrombotic Thrombocytopenic Purpura (TTP)
2. Haemolytic Uraemic Syndrome (HUS)
Typical HUS (STEC-HUS): Mainly supportive care — fluid management, renal replacement therapy if required. Avoid antimotility agents. Antibiotics are generally NOT recommended as they may increase Shiga toxin release. Monitor for haemolytic anaemia and AKI. Most children recover with supportive care alone.
3. Disseminated Intravascular Coagulation (DIC)
4. Malignant Hypertension
Control of blood pressure is the primary intervention. In the acute setting, IV labetalol or sodium nitroprusside infusion under arterial line monitoring targets a 25% reduction in mean arterial pressure over the first 24–48 hours (not rapid normalisation, to avoid cerebral hypoperfusion). Oral antihypertensives (amlodipine, perindopril) are transitioned once the patient stabilises. The MAHA resolves with blood pressure control.
5. Drug-Associated TMA
Immediate withdrawal of the offending agent is the cornerstone of management. Common culprits include quinine, cyclosporin, tacrolimus, clopidogrel, gemcitabine, bevacizumab, and VEGF tyrosine kinase inhibitors (sunitinib, sorafenib). ADAMTS13 levels may be normal or only mildly reduced. Supportive care including dialysis for AKI may be required. Report to the TGA via the Adverse Event Management System (AEMS).
Monitoring
Serial laboratory monitoring is essential during treatment and remission. The following parameters should be tracked:
| Parameter | Frequency | Target / Significance |
|---|---|---|
| Platelet count | Daily during acute TPE; twice weekly during taper | Sustained >150 × 10⁹/L for ≥2 days indicates remission in TTP |
| LDH | Daily during acute phase | Falling LDH correlates with disease response; rising LDH signals relapse |
| Haptoglobin | Every 2–3 days | Normalisation indicates cessation of haemolysis |
| Schistocyte count | Daily blood film review | Resolution of schistocytes supports clinical remission |
| ADAMTS13 activity | Weekly during treatment; at 1, 3, 6, 12 months post-remission | Persistent <10% predicts relapse; rising >20% indicates haematological remission |
| Renal function | Daily in HUS; as indicated in DIC/TTP | Improving eGFR in HUS; initiate RRT if oliguria/anuria persists |
| Coagulation studies | Every 6–12 hours in DIC; adjust with blood product support | Normalising PT, aPTT, fibrinogen, D-dimer indicate DIC resolution |
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836–2846.
- 2. Bendapudi PK, Hurwitz S, Fry A, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):e157–e164.
- 3. Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312–322.
- 4. Fakhouri F, Zuber J, Frémeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet. 2017;390(10095):681–696.
- 5. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol. 2009;145(1):24–33.
- 6. National Blood Authority Australia. Australian & New Zealand Society of Blood Transfusion Guidelines for the Administration of Blood Products. 3rd ed. Canberra: NBA; 2023.
- 7. Al-Horani RA. Potential anti-thrombotic therapies for thrombotic thrombocytopenic purpura. Cardiovasc Hematol Agents Med Chem. 2020;18(1):8–16.
- 8. Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. 2017;1(10):590–600.
- 9. Australian Institute of Health and Welfare. Aboriginal and Torres Strait Islander Health Performance Framework 2020 Summary Report. Canberra: AIHW; 2020.
- 10. RACGP. Red Book: Guidelines for Preventive Activities in General Practice. 10th ed. Melbourne: RACGP; 2018.
- 11. Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33(6):508–530.
- 12. Skerka C, Zipfel PF, Müller D, et al. The autoimmune disease DEAP-hemolytic uremic syndrome. Semin Thromb Hemost. 2010;36(6):625–632.