📋 Key Information Summary
- Waldenström macroglobulinaemia (WM) is a rare indolent lymphoplasmacytic lymphoma (LPL) characterised by bone marrow infiltration with clonal lymphoplasmacytic cells and secretion of monoclonal immunoglobulin M (IgM).
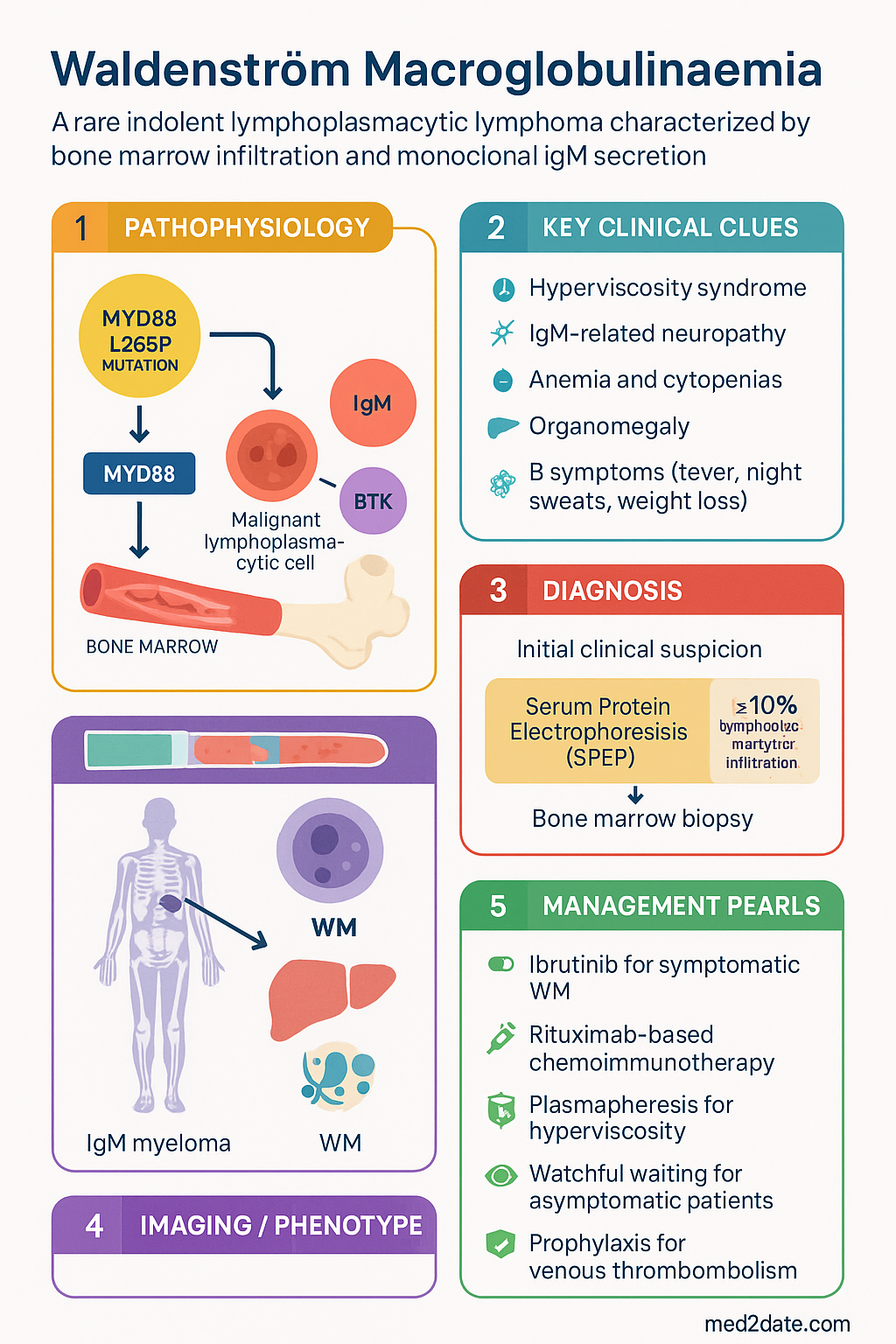
- The MYD88 L265P somatic mutation is present in >90% of WM cases and is a key diagnostic and potentially therapeutic target; CXCR4 WHIM-like mutations are found in ~30–40% and confer resistance to certain agents.
- Hyperviscosity syndrome is an oncological emergency — serum viscosity >4 cp or symptomatic patients require urgent plasmapheresis before chemotherapy.
- IgM-related neuropathy affects up to 20% of patients and may be the presenting feature; anti-MAG antibodies are implicated in distal sensory demyelinating neuropathy.
- Diagnosis requires bone marrow biopsy demonstrating ≥10% lymphoplasmacytic infiltration, immunophenotypic confirmation (CD19+, CD20+, CD5−, CD10−, CD23−), and identification of serum IgM monoclonal protein on serum protein electrophoresis (SPEP) and immunofixation.
- Ibrutinib (Imbruvica®) — a Bruton's tyrosine kinase inhibitor — is a first-line option for symptomatic WM, with or without rituximab; PBS Authority Required listing in Australia.
- Rituximab (MabThera®) is a cornerstone of combination therapy; commonly paired with cyclophosphamide, dexamethasone (DRC) or bendamustine (BR) for chemoimmunotherapy.
- Asymptomatic patients with low-level IgM and no cytopenias or hyperviscosity may be observed with watchful waiting.
- Treatment indications include symptomatic hyperviscosity, progressive cytopenias (Hb <100 g/L or platelets <100 × 10⁹/L), bulky disease, severe neuropathy, or cryoglobulinaemia.
- Venous thromboembolism risk is increased; IPCs and LMWH prophylaxis should be considered during immobility and chemotherapy.
- Incidence in Australia is approximately 0.5–0.6 per 100,000 population; median age at diagnosis 65–70 years, with a slight male predominance.
- Aboriginal and Torres Strait Islander peoples may present later due to access barriers; culturally safe, team-based care and health-literacy-appropriate education are essential.
Introduction & Australian Epidemiology
Waldenström macroglobulinaemia (WM) is a rare, indolent B-cell lymphoproliferative disorder classified as a lymphoplasmacytic lymphoma (LPL) by the World Health Organization (WHO). It is defined by the presence of bone marrow infiltration by clonal lymphoplasmacytic cells and evidence of an immunoglobulin M (IgM) monoclonal gammopathy. First described by Jan Gösta Waldenström in 1944, the disease occupies a unique niche between multiple myeloma and low-grade lymphomas.
In Australia, WM accounts for approximately 1–2% of all haematological malignancies. Cancer Australia data and state cancer registries suggest an age-standardised incidence of 0.5–0.6 per 100,000 persons per year, with approximately 150–200 new diagnoses annually. The median age at presentation is 65–70 years, and the male-to-female ratio is approximately 1.5:1. The disease is rare before age 40 and is slightly more common in individuals of Northern European descent.
The disease course is variable: many patients remain asymptomatic for years with stable IgM levels, while others develop progressive cytopenias, hyperviscosity syndrome, organomegaly, or debilitating neuropathy. Treatment decisions are guided by the presence of symptoms rather than IgM level alone. The landscape of WM management has been transformed over the past decade by targeted therapies, particularly Bruton's tyrosine kinase (BTK) inhibitors, and Australian patients have benefited from Pharmaceutical Benefits Scheme (PBS) listings of key agents.
Pathogenesis — MYD88 L265P Mutation
The molecular hallmark of WM is the somatic MYD88 L265P mutation, present in >90% of cases. MYD88 is an adaptor protein in the toll-like receptor (TLR) and interleukin-1 receptor (IL-1R) signalling pathways. The L265P gain-of-function mutation leads to constitutive activation of nuclear factor kappa B (NF-κB) through the IRAK1/IRAK4 complex, promoting malignant lymphoplasmacytic cell survival and proliferation.
Key Molecular Features
- MYD88 L265P: Found in >95% of WM cases. Constitutively activates BTK via the MYD88→IRAK1/4→NF-κB axis. This interaction is the basis for the efficacy of ibrutinib, which binds BTK at the Cys481 residue.
- CXCR4 WHIM-like mutations: Present in 30–40% of WM patients, predominantly in those with MYD88 L265P. These gain-of-function mutations increase CXCR4 surface expression and downstream AKT/ERK signalling, conferring resistance to ibrutinib and associated with higher IgM levels and symptomatic hyperviscosity.
- ARID1A, CD79B, TP53: Secondary mutations that may contribute to disease progression, transformation to aggressive lymphoma, or treatment resistance.
- Epigenetic dysregulation: Aberrant DNA methylation patterns and histone modification contribute to gene silencing and are under active investigation as therapeutic targets.
Pathophysiology of IgM-Related Complications
Monoclonal IgM exerts its pathological effects through several mechanisms:
- Hyperviscosity: IgM pentamers are the largest immunoglobulin class (~950 kDa). They aggregate red blood cells (rouleaux formation), increase serum viscosity, and impair microcirculatory blood flow, leading to neurological and haemorrhagic complications.
- Anti-myelin-associated glycoprotein (anti-MAG) neuropathy: IgM antibodies bind MAG on Schwann cell surfaces, disrupting myelin structure and causing a distal symmetric demyelinating sensorimotor neuropathy.
- Cryoglobulinaemia: Type I cryoglobulins (monoclonal IgM) precipitate at low temperatures, causing Raynaud phenomenon, skin ulceration, and glomerulonephritis.
- Amyloidosis (AL type): Rarely, IgM light chains form amyloid fibrils, depositing in kidneys, heart, nerves, and soft tissues.
- Cold agglutinin disease: IgM with anti-I specificity causes complement-mediated haemolysis, particularly in the cold.
Clinical Features — Hyperviscosity & Neuropathy
Up to 25% of WM patients are asymptomatic at diagnosis, detected incidentally through serum protein electrophoresis (SPEP). Symptomatic patients may present with one or more of the following syndromes:
Hyperviscosity Syndrome
Peripheral Neuropathy
IgM-related neuropathy affects 15–20% of WM patients at diagnosis and up to 50% during the disease course. Two main patterns are recognised:
- Anti-MAG neuropathy (most common): Distal, symmetric, predominantly sensory demyelinating neuropathy. Patients present with numbness, tingling, and gait unsteadiness, often beginning in the lower limbs. Anti-MAG antibodies are detectable in ~50% of WM neuropathy cases. Deep tendon reflexes are diminished, and sensory ataxia may be prominent.
- Axonal neuropathy: Less common; may be related to direct endoneurial deposition of IgM or amyloid. Presents with weakness and pain alongside sensory symptoms.
Other Presentations
- Cytopenias: Anaemia (Hb <100 g/L) is the most common finding at diagnosis. Thrombocytopenia and neutropaenia result from marrow infiltration or autoimmune mechanisms.
- Organomegaly: Hepatomegaly (20%), splenomegaly (15%), and lymphadenopathy (15%) are less prominent than in other lymphomas.
- B symptoms: Fever, night sweats, and unexplained weight loss occur in 10–15%.
- Bing–Neel syndrome: Direct CNS infiltration by malignant lymphoplasmacytic cells causing confusion, cranial nerve palsies, or spinal cord compression. A rare but devastating complication.
- Renal involvement: Light-chain deposition disease, cryoglobulinaemic glomerulonephritis, or amyloidosis may cause nephrotic syndrome or renal impairment.
- Skin: Schnitzler syndrome (chronic urticaria, monoclonal IgM, fever, arthralgia, bone pain) and cold-agglutinin-related acrocyanosis.
Investigations — SPEP, Bone Marrow Biopsy & Beyond
Diagnostic Criteria (IPWMWS / WHO 2022)
The diagnosis of WM requires all of the following:
- Any amount of serum IgM monoclonal protein (detected by SPEP and immunofixation)
- Bone marrow biopsy showing ≥10% infiltration by small lymphocytes, plasmacytoid lymphocytes, and plasma cells with an interstitial, nodular, or diffuse pattern
- Immunophenotypic evidence of LPL: CD19+, CD20+, surface IgM+, CD5−, CD10−, CD23− (typically; variations occur)
- Absence of features diagnostic of other lymphomas or multiple myeloma
Essential Investigations
Risk Stratification & Prognostic Scoring
International Prognostic Scoring System for WM (IPSSWM / Revised IPSSWM)
| Risk Group | Criteria | Median OS |
|---|---|---|
| Low risk | Age ≤65 AND β2-microglobulin <3 mg/L AND Hb ≥115 g/L | Not reached (>15 years) |
| Intermediate risk | Any factor: age >65, β2-microglobulin 3–5.5 mg/L, Hb <115 g/L (but not high risk) | ~9–12 years |
| High risk | β2-microglobulin >5.5 mg/L | ~3–5 years |
Additional Prognostic Factors
- CXCR4 mutations: Associated with higher IgM levels, increased hyperviscosity risk, and reduced response to ibrutinib monotherapy.
- TP53 mutations/deletion: Predict poorer outcomes with chemoimmunotherapy; may favour targeted therapy approaches.
- LDH elevation: May indicate transformation to aggressive lymphoma (DLBCL).
- IgM >60 g/L: Higher risk of hyperviscosity; monitor closely even if asymptomatic.
Management — Ibrutinib, Rituximab & Plasmapheresis
Treatment Indications
Treatment should be initiated for symptomatic disease. Watchful waiting is appropriate for asymptomatic patients with low-level IgM and preserved blood counts.
- Symptomatic hyperviscosity
- Progressive cytopenias (Hb <100 g/L or platelets <100 × 10⁹/L) due to marrow infiltration
- Moderate-to-severe neuropathy (functional impairment)
- Bulky or symptomatic lymphadenopathy / organomegaly
- Systemic symptoms (B symptoms) attributable to WM
- Symptomatic cryoglobulinaemia or cold agglutinin disease
- AL amyloidosis
- Rapidly rising IgM (doubling time <6 months)
Emergency Management — Plasmapheresis
Targeted Therapy — BTK Inhibitors
Monoclonal Antibody — Rituximab
Chemoimmunotherapy Regimens
| Regimen | Components | Cycle | Notes |
|---|---|---|---|
| DRC | Dexamethasone 20 mg PO D1–4, Rituximab 375 mg/m² IV D1, Cyclophosphamide 100 mg/m² PO D1–5 | Every 21 days × 6 cycles | Well-tolerated first-line option. PBS-listed components. |
| BR | Bendamustine 90 mg/m² IV D1–2, Rituximab 375 mg/m² IV D1 | Every 28 days × 6 cycles | Higher overall response rate (~95%) vs DRC. More myelosuppression. PBS Authority Required for bendamustine. |
| Ibrutinib + Rituximab | Ibrutinib 420 mg PO daily + Rituximab 375 mg/m² IV weekly × 4 then monthly × 4 | Continuous ibrutinib; rituximab as above | iNNOVATE trial showed superior PFS vs rituximab alone. Preferred for elderly/comorbid patients. Avoid in patients with uncontrolled AF. |
| Bortezomib-based | Bortezomib 1.6 mg/m² IV D1, 8, 15 + Dexamethasone 20 mg + Rituximab 375 mg/m² | Every 28 days × 6 cycles | Rapid response; useful for urgent IgM reduction. Peripheral neuropathy risk limits duration. PBS Authority Required. |
Other Agents
Treatment Algorithm
Monitoring
Response Assessment (IWWM Consensus Criteria)
| Response | Criteria |
|---|---|
| Complete Response (CR) | Negative immunofixation, no histological evidence of disease, resolution of extramedullary disease |
| Very Good Partial Response (VGPR) | ≥90% reduction in serum IgM from baseline |
| Partial Response (PR) | 50–89% reduction in serum IgM from baseline |
| Minor Response (MR) | 25–49% reduction in serum IgM from baseline |
| Stable Disease (SD) | <25% change in IgM, no new disease features |
| Progressive Disease (PD) | ≥25% increase in IgM (confirmed × 2, ≥12 weeks apart) or new cytopenias, organomegaly, or hyperviscosity |
Follow-Up Schedule
- During active treatment: FBC, serum IgM, viscosity every 2–4 weeks. LFTs, renal function each cycle.
- On maintenance / post-treatment: FBC, serum IgM, viscosity every 3 months for 2 years, then every 6 months if stable.
- Annual: Bone marrow biopsy if deep response assessment needed (e.g., for clinical trial or CR assessment). CT or PET-CT if extramedullary disease was present.
- On ibrutinib/zanubrutinib: ECG at baseline and if symptoms of AF develop. Blood pressure monitoring at each visit. Full blood count for cytopenias. Screen for second malignancies annually.
- Neuropathy: Serial NCS every 6–12 months if anti-MAG positive. Functional assessment (INCAT disability score).
Special Populations
Pregnancy
Paediatrics
Elderly (≥75 years)
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenström's macroglobulinaemia: consensus panel recommendations from the Second International Workshop on Waldenström's Macroglobulinaemia. Semin Oncol. 2003;30(2):110-115.
- 2. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström's macroglobulinaemia. N Engl J Med. 2012;367(9):826-833.
- 3. Treon SP, Cao Y, Xu L, et al. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinaemia. Blood. 2014;123(18):2791-2796.
- 4. Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenström's macroglobulinemia. N Engl J Med. 2018;378(25):2399-2410.
- 5. Tam CS, Opat S, D'Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038-2050.
- 6. Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström's Macroglobulinaemia. Blood. 2016;128(10):1321-1328.
- 7. Kyle RA, Treon SP, Alexanian R, et al. Prognostic markers and criteria to initiate therapy in Waldenström's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenström's Macroglobulinemia. Semin Oncol. 2003;30(2):116-120.
- 8. Australian Institute of Health and Welfare (AIHW). Cancer data in Australia. Canberra: AIHW; 2024. Available from: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia
- 9. Riva M, Ferreri AJM, Motta V, et al. Bing-Neel syndrome: an update. Haematologica. 2017;102(2):e58-e60.
- 10. Department of Health and Aged Care. Pharmaceutical Benefits Scheme: ibrutinib. Canberra: Australian Government; 2024. Available from: https://www.pbs.gov.au/
- 11. National Health and Medical Research Council (NHMRC). National statement on ethical conduct in human research. Canberra: NHMRC; 2023.
- 12. Kastritis E, Leblond V, Dimopoulos MA, et al. Waldenström's macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(7):1104-1115.