📋 Key Information Summary
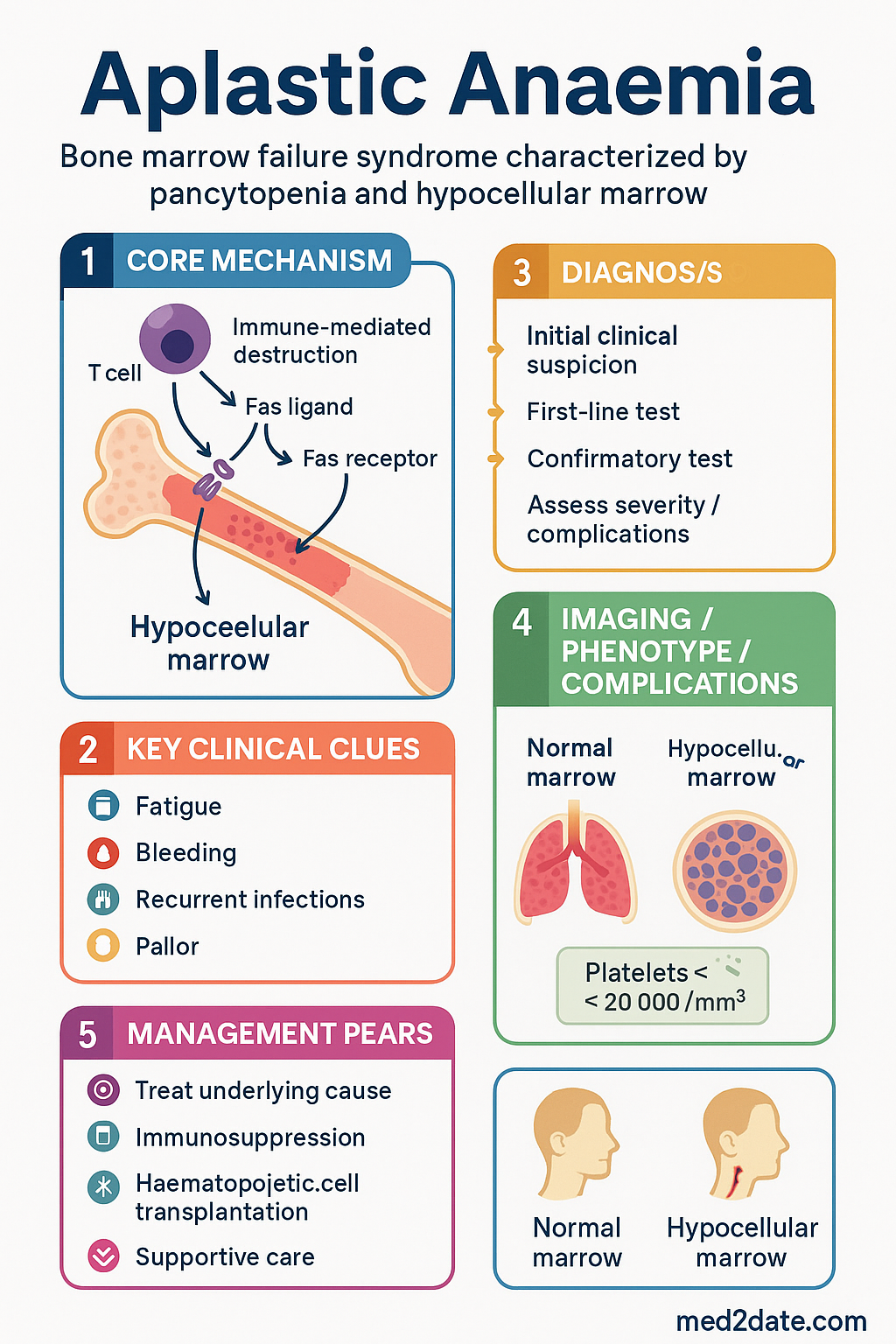
- Aplastic anaemia (AA) is a bone marrow failure syndrome characterised by pancytopenia and a hypocellular marrow, resulting from immune-mediated destruction of haematopoietic stem cells.
- Approximately 65–75% of cases are idiopathic (immune-mediated); known causes include drugs (chloramphenicol, carbimazole, sulphonamides), viruses (hepatitis, parvovirus B19, EBV), and inherited bone marrow failure syndromes.
- Severity classification (Camitta/modified Camitta criteria) guides treatment: non-severe AA, severe AA (SAA), and very severe AA (vSAA) based on reticulocyte count, neutrophil count, and platelet count.
- Incidence in Australia is approximately 2–3 per million population per year; higher rates observed in Aboriginal and Torres Strait Islander populations in remote areas.
- Urgent referral to a haematology centre is required for all patients with SAA or vSAA — do not delay while awaiting investigations.
- Allogeneic haematopoietic stem cell transplantation (allo-HSCT) from an HLA-matched sibling donor is the treatment of choice for patients aged <40 years with SAA/vSAA.
- Immunosuppressive therapy (IST) with horse anti-thymocyte globulin (hATG) plus ciclosporin is first-line for patients aged >40 years or without a matched sibling donor.
- Eltrombopag (thrombopoietin receptor agonist) combined with IST significantly improves response rates and is now standard of care alongside hATG + ciclosporin.
- Horse ATG (Atgam®) is preferred over rabbit ATG in Australia for first-line IST based on superior response and survival data.
- Transfusion support with irradiated, CMV-safe blood products is essential during workup and treatment; iron chelation is needed if ferritin >1000 μg/L.
- Bone marrow biopsy is mandatory to confirm diagnosis and exclude myelodysplastic syndrome (MDS), paroxysmal nocturnal haemoglobinuria (PNH), and inherited marrow failure syndromes.
- HLA typing and donor search should be initiated at diagnosis for all patients aged <50 years.
- G-CSF is not recommended as a bridging measure in SAA; it does not improve outcomes and may delay definitive therapy.
- All patients require lifelong haematology follow-up; relapse after IST occurs in 15–25% of responders.
- Aboriginal and Torres Strait Islander patients may have delayed presentation due to healthcare access barriers and require culturally safe care coordination with RFDS and regional haematology services.
Introduction & Australian Epidemiology
Aplastic anaemia is a rare, potentially life-threatening bone marrow failure syndrome characterised by peripheral blood pancytopenia and a hypocellular bone marrow. The underlying mechanism in the majority of acquired cases is immune-mediated destruction of pluripotent haematopoietic stem and progenitor cells by autoreactive cytotoxic T lymphocytes, leading to an empty or near-empty marrow.
In Australia, the age-standardised incidence is approximately 2–3 per million population per year, consistent with Western population data. The disease demonstrates a bimodal age distribution with peaks in young adults (15–25 years) and older adults (>60 years). There is no significant sex predilection in idiopathic cases, though drug- and toxin-associated aplasia may show occupational or exposure-related patterns.
The Australian health system context presents both advantages and challenges. Major haematology and transplant centres in Sydney, Melbourne, Brisbane, Perth, and Adelaide provide specialised care including allo-HSCT. However, patients in rural, remote, and very remote areas — including many Aboriginal and Torres Strait Islander communities — face significant delays in diagnosis and access to tertiary haematology services. The Royal Flying Doctor Service (RFDS) and telehealth haematology consultations play a critical role in bridging this gap.
Without treatment, severe aplastic anaemia carries a mortality rate exceeding 70% within 12 months due to infection or haemorrhage. With modern therapy — including matched sibling transplant and combined IST with eltrombopag — overall survival at 5 years is 75–90%. Early recognition, urgent referral, and appropriate treatment selection are therefore critical.
Aetiology
Aplastic anaemia may be inherited or acquired. The aetiology determines both the approach to investigation and, in some cases, the treatment strategy. In approximately 65–75% of cases, no identifiable cause is found and the condition is classified as idiopathic, though immune-mediated mechanisms are strongly implicated.
Idiopathic (Immune-Mediated)
The majority of acquired aplastic anaemia cases are idiopathic and considered immune-mediated. Autoreactive cytotoxic T lymphocytes (CD8+ T cells) targeting haematopoietic stem cells are identified in the bone marrow of most patients. Clonal expansion of these T cells, often oligoclonal, leads to targeted destruction of stem cells via Fas-ligand and perforin/granzyme pathways. Cytokine-mediated suppression (TNF-α, IFN-γ) further inhibits residual haematopoiesis.
This immune basis explains the response to immunosuppressive therapy and the occasional co-existence of aplastic anaemia with other autoimmune conditions including thyroiditis, systemic lupus erythematosus, and thymoma.
Drug-Associated
A number of medications have been associated with aplastic anaemia, either through dose-dependent myelosuppression or idiosyncratic immune-mediated reactions:
| Drug Category | Examples | Mechanism | Reversibility |
|---|---|---|---|
| Dose-dependent myelotoxins | Cytotoxic chemotherapy agents (alkylators, antimetabolites), benzene | Direct stem cell toxicity | Usually reversible on cessation |
| Idiosyncratic (dose-independent) | Chloramphenicol, carbimazole, methimazole, sulphonamides, NSAIDs (phenylbutazone, indomethacin), gold salts, D-penicillamine, allopurinol | Immune-mediated or direct toxicity with genetic susceptibility | May be irreversible despite drug withdrawal |
| Anticonvulsants | Carbamazepine, phenytoin, sodium valproate | Idiosyncratic immune-mediated | Usually reversible, but may persist |
| Antibiotics | Co-trimoxazole, flucloxacillin (rare) | Idiosyncratic | Variable |
Viral-Associated
Several viral infections have been implicated in aplastic anaemia, either through direct marrow suppression or immune-mediated mechanisms:
- Hepatitis viruses: Hepatitis-associated aplastic anaemia (HAAA) accounts for approximately 5% of cases. Typically occurs 2–3 months after acute hepatitis, often seronegative hepatitis (non-A to E). Mortality is high without treatment. HAAA is particularly important in younger patients.
- Parvovirus B19: Causes selective red cell aplasia rather than true pancytopenic aplastic anaemia, though severe immunodeficiency may lead to broader marrow suppression.
- Epstein-Barr virus (EBV): Rare association with aplastic anaemia, usually in the context of severe EBV infection or post-transplant lymphoproliferative disease.
- HIV: Direct marrow infection and opportunistic infections (CMV, mycobacteria) can cause marrow failure; distinguish from HIV-associated cytopenias.
- Cytomegalovirus (CMV): Particularly relevant in immunocompromised patients and post-transplant settings.
Inherited Bone Marrow Failure Syndromes
Inherited causes must be considered especially in children, young adults, and those with a family history or associated physical anomalies:
- Fanconi anaemia: Autosomal recessive or X-linked; chromosomal breakage test (diepoxybutane or mitomycin C) is diagnostic. Associated with short stature, radial ray anomalies, skin pigmentation, and predisposition to MDS/AML.
- Dyskeratosis congenita: Telomere biology disorder; mucosal leukoplakia, nail dystrophy, skin pigmentation. TERC and TERT gene mutations.
- Shwachman-Diamond syndrome: Exocrine pancreatic insufficiency, skeletal anomalies, neutropenia progressing to aplastic anaemia.
- 先天性角化不良 (DC) and other telomere biology disorders may present in adulthood and should be considered in apparently acquired AA, as IST may be less effective.
Clinical Features & Severity Classification
Clinical Presentation
The clinical presentation of aplastic anaemia reflects the consequences of pancytopenia. Onset is typically insidious over weeks to months, though hepatitis-associated cases may present more acutely:
Other clinical features may include mild hepatosplenomegaly (rare in idiopathic AA; if present, consider PNH or MDS), lymphadenopathy (should prompt consideration of lymphoma or viral infection), and congenital anomalies suggesting inherited marrow failure syndrome.
Severity Classification
Severity classification is essential for treatment decisions and is based on peripheral blood counts and bone marrow cellularity. The modified Camitta criteria are used internationally and in Australian practice:
| Parameter | Non-Severe AA | Severe AA (SAA) | Very Severe AA (vSAA) |
|---|---|---|---|
| Bone marrow cellularity | <25% (or 25–50% with <30% haematopoietic cells) | <25% | <25% |
| Reticulocyte count | ≥20 × 10⁹/L | <20 × 10⁹/L (<1%) | <20 × 10⁹/L (<1%) |
| Neutrophil count (ANC) | ≥0.5 × 10⁹/L | <0.5 × 10⁹/L | <0.2 × 10⁹/L |
| Platelet count | ≥20 × 10⁹/L | <20 × 10⁹/L | <20 × 10⁹/L |
Clonal Evolution
Approximately 10–15% of patients with aplastic anaemia may evolve to myelodysplastic syndrome (MDS) or acute myeloid leukaemia (AML) over 10 years. Monosomy 7 and trisomy 8 are the most common cytogenetic abnormalities detected at clonal evolution. PNH clones are detectable in 40–50% of patients at diagnosis and may expand over time; however, progression to clinically significant haemolysis occurs in a minority.
Investigations
A systematic approach to investigation is essential to confirm the diagnosis, assess severity, exclude secondary causes, and guide treatment selection. Bone marrow biopsy is the cornerstone diagnostic investigation.
Baseline Blood Investigations
Bone Marrow Biopsy
Bone marrow biopsy findings in aplastic anaemia include:
- Cellularity: Markedly hypocellular marrow (<25% cellularity for age). In patients under 60 years, normal cellularity is 30–70%; thus <25% is clearly abnormal.
- Haematopoietic cells: Severely reduced trilineage haematopoiesis. Residual haematopoietic islands may be seen.
- Stroma: Predominantly fat and stromal cells. No increase in blasts (<5%). No significant fibrosis (reticulin stain should be ≤MF-1).
- Trephine immunohistochemistry: CD34+ cells markedly reduced. Useful to exclude MDS (dysplastic megakaryocytes) and lymphoproliferative infiltrates.
- Cytogenetics: Conventional karyotyping on bone marrow aspirate. Clonal cytogenetic abnormalities (monosomy 7, trisomy 8) suggest coexisting MDS.
Additional Specialised Investigations
Differential Diagnosis to Exclude
| Diagnosis | Distinguishing Features | Key Investigation |
|---|---|---|
| Myelodysplastic syndrome (MDS) | Dysplastic morphology, ring sideroblasts, clonal cytogenetics | BM trephine morphology, karyotype, NGS panel |
| Paroxysmal nocturnal haemoglobinuria (PNH) | Haemolysis, thrombosis, large PNH clone | FLAER flow cytometry |
| Acute leukaemia | Blasts ≥20% in marrow | BM aspirate morphology, flow cytometry |
| Hypersplenism | Splenomegaly, marrow hypercellular | Ultrasound, BM biopsy |
| Vitamin B12/folate deficiency | Macrocytic anaemia, megaloblastic changes | Serum B12, folate, homocysteine, MMA |
| Inherited marrow failure | Congenital anomalies, family history, young age | Chromosomal breakage, telomere length, genetic testing |
Management
Treatment of aplastic anaemia depends on disease severity, patient age, donor availability, and comorbidities. The three principal treatment modalities are allogeneic haematopoietic stem cell transplantation (allo-HSCT), immunosuppressive therapy (IST), and supportive care. Eltrombopag has transformed IST outcomes and is now integrated into first-line regimens.
Treatment Algorithm
Allogeneic Haematopoietic Stem Cell Transplantation (allo-HSCT)
Allo-HSCT from an HLA-matched sibling donor (MSD) is the treatment of choice for patients aged <40 years with SAA or vSAA, offering cure rates of 75–90%. Transplant should be performed as soon as possible after diagnosis, ideally before the patient becomes heavily transfused (which increases rejection and GVHD risk).
Key transplant considerations in Australia:
- All blood products must be irradiated and CMV-safe (leucodepleted, CMV-seronegative or equivalent) from diagnosis onwards.
- Red cell and platelet transfusions should be antigen-matched where possible (especially for red cells) to reduce alloimmunisation.
- For patients aged 40–50 years with an MSD, reduced-intensity conditioning regimens are increasingly used with acceptable outcomes. Decisions should be made at a multidisciplinary transplant meeting.
- Unrelated donor HSCT is considered second-line (after IST failure) or first-line if a well-matched unrelated donor is rapidly available. Outcomes have improved with high-resolution HLA matching through the Australian Bone Marrow Donor Registry (ABMDR).
- Cord blood transplant is an alternative when no matched donor is available, though engraftment rates are lower.
Immunosuppressive Therapy (IST)
IST is first-line treatment for patients aged >40 years without a matched sibling donor, and for younger patients awaiting a donor or with comorbidities precluding transplant. The combination of horse ATG (hATG) with ciclosporin and eltrombopag is the current standard of care.
Second-Line Therapy
For patients who fail to respond to first-line IST (no response at 4–6 months) or who relapse after initial response, second-line options include:
- Repeat IST with rabbit ATG + ciclosporin + eltrombopag: Response rate approximately 30–40% for second course. Preferred if patient is not a transplant candidate.
- Allo-HSCT from unrelated donor: Consider for younger patients (<50 years) with an available well-matched donor. Outcomes have improved significantly with high-resolution HLA matching.
- Eltrombopag monotherapy: May be used in IST-refractory patients not eligible for transplant. Response rate approximately 40% in this setting.
- Haploidentical HSCT: Emerging option using post-transplant cyclophosphamide for GVHD prophylaxis. Outcomes approaching MSD transplant in selected centres.
Supportive Care
Supportive care is essential for all patients, regardless of definitive therapy:
- All blood products must be irradiated (prevent transfusion-associated GVHD) and CMV-safe.
- Red cell transfusion threshold: maintain Hb >70 g/L (or >80 g/L if symptomatic cardiovascular disease).
- Platelet transfusion threshold: <10 × 10⁹/L prophylactic; <20 × 10⁹/L with fever; any count if active bleeding.
- Antigen-matched red cells where possible (extended phenotype matching).
- Iron chelation (deferasirox 10–20 mg/kg/day PO) when ferritin >1000 μg/L and ongoing transfusion requirement.
- Neutropenic precautions when ANC <0.5 × 10⁹/L.
- Antibacterial prophylaxis: ciprofloxacin 500 mg PO BD if ANC <0.5 × 10⁹/L (per eTG guidelines).
- Antifungal prophylaxis: fluconazole 200 mg PO daily; escalate to posaconazole if prolonged profound neutropenia or HSCT.
- Antiviral prophylaxis: aciclovir 400 mg PO BD (HSV/VZV prophylaxis).
- PJP prophylaxis: co-trimoxazole 480 mg PO daily or 960 mg PO 3 times/week if on IST (use cautiously — may worsen cytopenias).
- Vaccination review — live vaccines contraindicated during IST and for 6 months after.
Monitoring Response to IST
Special Populations
🇦🇺 Aboriginal and Torres Strait Islander Health Considerations
Aplastic anaemia in Aboriginal and Torres Strait Islander peoples requires particular attention to healthcare access, cultural safety, and the unique epidemiological and social context of Indigenous Australian communities. Data from the AIHW and jurisdictional cancer registries suggest that while overall incidence may be similar to the non-Indigenous population, outcomes are often poorer due to delayed diagnosis and barriers to accessing specialist care.
📚 References
- 1. Patel BA, Groarke EM, Lotter J, et al. Long-term outcomes in patients with severe aplastic anaemia treated with immunosuppression and eltrombopag: a phase 2 study. Lancet Haematol. 2022;9(11):e804–e814. doi:10.1016/S2352-3026(22)00282-X
- 2. Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376(16):1540–1550. doi:10.1056/NEJMoa1613878
- 3. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129(11):1428–1436. doi:10.1182/blood-2016-08-693689
- 4. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172(2):187–207. doi:10.1111/bjh.13853
- 5. Peffault de Latour R, Tabbara IA, Bhatt VR, et al. Allogeneic transplantation for aplastic anemia. Biol Blood Marrow Transplant. 2020;26(8):e162–e169. doi:10.1016/j.bbmt.2020.04.022
- 6. Marsh JCW, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70. doi:10.1111/j.1365-2141.2009.07842.x
- 7. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120(6):1185–1196. doi:10.1182/blood-2011-12-274019
- 8. Australian Bone Marrow Donor Recruit (ABMDR). Annual Report 2023. Sydney: ABMDR; 2023. Available from: www.abmdr.org.au
- 9. AIHW (Australian Institute of Health and Welfare). Blood and blood-forming organ diseases in Aboriginal and Torres Strait Islander peoples. Cat. no. IHW 195. Canberra: AIHW; 2021.
- 10. Australasian Society of Clinical Immunology and Allergy (ASCIA). Guidelines for the use of immunoglobulin and related therapies in haematological disorders. ASCIA Position Statement. 2023.
- 11. RHDAustralia. Australian guidelines for the prevention, diagnosis and management of rheumatic heart disease. 2nd edition. Darwin: RHDAustralia, Menzies School of Health Research; 2020.
- 12. Australian Commission on Safety and Quality in Health Care (ACSQHC). National Safety and Quality Health Service Standards. 2nd edition. Sydney: ACSQHC; 2021.