📋 Key Information Summary
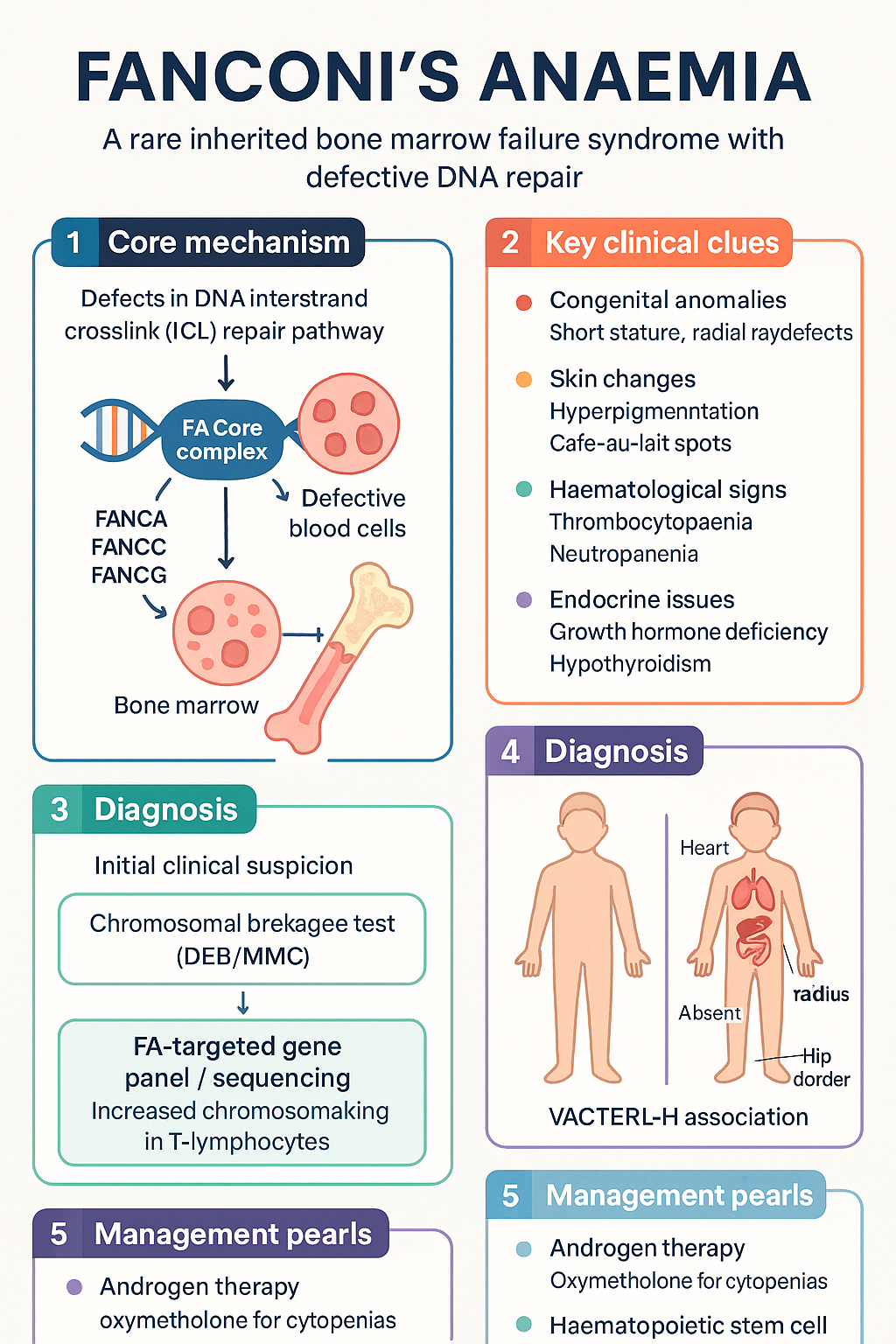
- Fanconi anaemia (FA) is a rare autosomal recessive (rarely X-linked) inherited bone marrow failure syndrome caused by defects in the DNA interstrand crosslink (ICL) repair pathway — at least 22 complementation groups (FANCA–FANCW) are now identified.
- Incidence is approximately 1 in 160,000 live births; carrier frequency estimated at 1 in 180–300 in outbred populations, with higher rates in founder populations (Ashkenazi Jewish, Afrikaner, Spanish Romani).
- Over 60% of patients present with congenital anomalies including short stature, radial ray defects, hyperpigmentation, microcephaly, renal malformations, and small gonads; up to 30% are phenotypically normal at birth.
- Progressive bone marrow failure affects ≥80% of patients by age 40, progressing through hypocellular myelodysplasia to aplastic anaemia.
- Chromosomal breakage test (diepoxybutane or mitomycin C-induced) is the gold-standard diagnostic assay — demonstrates increased radial figures and chromosomal breaks in phytohaemagglutinin-stimulated T-lymphocytes.
- Patients harbour a markedly elevated lifetime risk of acute myeloid leukaemia (AML) and solid tumours (head and neck squamous cell carcinoma, vulvar, oesophageal) — cumulative cancer incidence ~40% by age 50.
- Androgen therapy (oxymetholone ± G-CSF) is first-line pharmacological treatment for cytopenias; haematopoietic stem cell transplant (SCT) remains the only curative option for bone marrow failure and myelodysplasia/leukaemia.
- SCT outcomes are best with an HLA-matched sibling donor using reduced-intensity conditioning (RIC) regimens to minimise transplant-related mortality.
- Lifelong cancer surveillance is mandatory: annual ENT examination from age 10, gynaecological screening from menarche, and screening colonoscopy from age 35 (or 10 years post-SCT).
- All patients require genetic counselling; sibling screening with chromosomal breakage testing is essential.
- Oxymetholone is PBS-listed (Authority Required) for aplastic anaemia in Australia; G-CSF (filgrastim) is PBS-listed as General Benefit for congenital neutropaenia and bone marrow failure.
- Aboriginal and Torres Strait Islander patients may present later due to reduced access to specialist and genetic services; culturally appropriate engagement and remote telehealth support are essential.
- Female patients require gynaecological and obstetric input — gonadal insufficiency is near-universal; oestrogen replacement is indicated for delayed puberty and bone protection.
Introduction & Australian Epidemiology
Fanconi anaemia (FA) is a rare, clinically heterogeneous inherited bone marrow failure syndrome characterised by defective DNA interstrand crosslink (ICL) repair. First described by Swiss paediatrician Guido Fanconi in 1927, FA is now understood to arise from biallelic (or, in the case of FANCB, hemizygous X-linked) pathogenic variants in one of at least 22 DNA repair genes that converge on a common pathway for resolving interstrand crosslinks and maintaining genomic stability.
FA has a worldwide incidence of approximately 1 in 160,000 live births, with an estimated heterozygous carrier frequency of 1 in 180–300 in outbred populations. Certain founder mutations substantially increase prevalence in specific populations: the Ashkenazi Jewish population (FANCC c.456+4A>T carrier frequency ~1 in 89), Afrikaner populations in South Africa (FANCG), and Spanish Romani communities (FANCA).
In Australia, FA accounts for a significant proportion of inherited bone marrow failure syndromes referred to tertiary paediatric haematology centres. The Royal Children's Hospital Melbourne, Sydney Children's Hospital, and the Women's and Children's Hospital Adelaide are the principal diagnostic and transplant centres. Australian data from the Australasian Bone Marrow Transplant Recipient Registry (ABMTRR) confirm approximately 2–4 SCT procedures for FA per year nationally. No specific Australian FA registry exists, though patients are captured by the ABMTRR and state-based genetics services.
The clinical course is dominated by progressive bone marrow failure, developmental anomalies, extreme cancer predisposition, and endocrine complications. Management requires a multidisciplinary team including paediatric haematology, genetics, endocrinology, ENT, gynaecology, and long-term adult survivorship care.
Genetics & DNA Repair Defect
The FA pathway is a complex signalling and repair network essential for resolving DNA interstrand crosslinks (ICLs) that would otherwise stall replication forks and cause chromosomal breakage. The pathway involves at least 22 complementation groups:
| Gene | Protein | Inheritance | Relative Frequency |
|---|---|---|---|
| FANCA | FANCA | AR | ~60–65% |
| FANCC | FANCC | AR | ~10–15% |
| FANCG | FANCG/XRCC9 | AR | ~10% |
| FANCD2 | FANCD2 | AR | ~3% |
| FANCJ/BRIP1 | BRIP1 | AR | ~2% |
| FANCB | FANCB | X-linked | ~2% |
| FANCD1/BRCA2 | BRCA2 | AR | ~2–3% |
| FANCN/PALB2 | PALB2 | AR | ~1–2% |
| Others (FANCL, FANCM, FANCO, FANCP, FANCQ, FANCR, FANCS, FANCT, FANCU, FANCV, FANCW) | Various | AR | ~5% combined |
Molecular Pathway
The FA core complex (FANCA, B, C, E, F, G, L, M, T) functions as an E3 ubiquitin ligase that monoubiquitinates the FANCI–FANCD2 (ID2) complex upon replication fork stalling at an ICL. Ubiquitinated ID2 recruits downstream effectors including FANCD1 (BRCA2), FANCJ (BRIP1), FANCN (PALB2), FANCO (RAD51C), FANCS (BRCA1), and FANCR (RAD51) for homologous recombination repair. Deficiency at any step leads to accumulation of unrepaired ICLs, chromosomal breakage, chromosomal instability, and cell death — particularly in haematopoietic stem and progenitor cells.
Inheritance & Genetic Counselling
- Autosomal recessive for all groups except FANCB (X-linked recessive — all affected individuals are male).
- Recurrence risk for siblings: 25% affected, 50% carrier, 25% unaffected non-carrier (for AR forms).
- Siblings of affected probands should undergo chromosomal breakage testing regardless of phenotype.
- Carrier testing and prenatal/pre-implantation genetic diagnosis (PGD) are available at Australian genetics centres (e.g., Victorian Clinical Genetics Services, NSW Health Pathology Genetics).
- Somatic mosaicism (revertant mosaicism) occurs in 10–25% of FA patients and may confound diagnostic testing.
Clinical Features & Congenital Anomalies
FA exhibits wide phenotypic variability even within families carrying identical mutations. Approximately 60–75% of patients have at least one congenital anomaly; however, up to 30% appear phenotypically normal at birth, leading to delayed diagnosis.
Congenital Anomalies
| System | Anomalies | Frequency |
|---|---|---|
| Skeletal — upper limb | Radial ray defects (hypoplastic/absent thumbs, radial aplasia), thenar hypoplasia | 40–50% |
| Skin | Generalised hyperpigmentation, café-au-lait spots, hypopigmented patches | 50–60% |
| Growth | Short stature (proportionate), low birth weight | 50–65% |
| Craniofacial | Microcephaly, micrognathia, small eyes, epicanthic folds, ear anomalies | 30–50% |
| Renal | Horseshoe kidney, renal agenesis, duplicated collecting system, renal hypoplasia | 20–30% |
| Genitourinary | Hypogonadism, hypospadias, undescended testes, bicornuate uterus, vaginal atresia | 15–30% |
| Eyes | Strabismus, cataracts, microphthalmia, ptosis | 10–25% |
| GI/hepatic | Oesophageal atresia, tracheo-oesophageal fistula, biliary atresia | 5–10% |
| Cardiac | VSD, ASD, PDA, coarctation of the aorta | 5–10% |
| CNS | Structural CNS malformations, developmental delay (mild) | 5–10% |
| Hearing | Sensorineural or conductive hearing loss | 5–10% |
VACTERL-H Association
FA is an important differential diagnosis in any child with features of VACTERL-H (Vertebral, Anal, Cardiac, Tracheo-Esophageal, Renal, Limb anomalies + Hydrocephalus). All children with radial ray anomalies or VACTERL-H should be screened with a chromosomal breakage test.
Haematological Manifestations
- Median age at diagnosis of bone marrow failure: 7 years (range birth to 40+ years).
- Thrombocytopaenia is often the first cytopenia detected, followed by neutropaenia and then anaemia.
- Progressive macrocytosis (elevated HbF, raised MCV) is an early haematological clue even before overt cytopenias.
- ≥80% of patients develop clinically significant bone marrow failure by age 40.
- Myelodysplastic syndrome (MDS) occurs in 20–30% and may progress to AML.
Endocrine Manifestations
- Growth hormone deficiency and hypothyroidism are common.
- Gonadal insufficiency: primary ovarian insufficiency in females; testicular failure in males.
- Insulin resistance and glucose intolerance develop in a significant proportion of adults.
- Reduced bone mineral density — compounded by chronic corticosteroid or androgen therapy.
Investigations (Chromosomal Fragility Test)
Diagnostic Testing
MBS Item Summary for Key Investigations
| Investigation | MBS Item | Notes |
|---|---|---|
| FBC + blood film | 65070 | Serial monitoring essential |
| Hb electrophoresis | 65095 | Elevated HbF for age |
| TSH, fT4 | 66647 | Screen at diagnosis, annually |
| IGF-1 | 66812 | GH deficiency screening |
| Echocardiogram | 55124 | Structural anomaly screening |
| Renal ultrasound | 55300 | Structural anomaly screening |
| MRI brain | 63001 | CNS anomaly screening |
Management (Androgens, SCT, Surveillance)
Androgen Therapy
Androgens stimulate erythropoiesis and can produce meaningful haematological responses in FA patients with cytopenias. They are first-line pharmacological therapy when SCT is not immediately indicated.
Haematopoietic Stem Cell Transplantation (SCT)
SCT is the only curative treatment for FA-related bone marrow failure and myelodysplasia/AML. However, FA cells are exquisitely sensitive to alkylating agents and ionising radiation, necessitating modified transplant conditioning regimens.
Transplant Indications
- Transfusion-dependent thrombocytopaenia or anaemia unresponsive to androgens.
- Severe neutropaenia (ANC <0.5 × 10⁹/L) with recurrent life-threatening infections.
- Progressive MDS with increasing blast count or adverse cytogenetics (monosomy 7, complex karyotype).
- Evolution to AML.
- Early transplant with a matched sibling donor before severe marrow failure may be considered in selected cases.
Cancer Surveillance Programme
FA patients have a cumulative lifetime cancer risk of approximately 40% by age 50. Head and neck squamous cell carcinoma (HNSCC) and anogenital squamous cell carcinoma are particularly over-represented, with risk further increased post-SCT due to conditioning-related mucosal damage and chronic GVHD.
Supportive Care
- Infection prevention: Irradiated, CMV-safe blood products for all transfusions (FA cells are hypersensitive to allogeneic lymphocytes). Leucodepleted products as standard. Pneumocystis jirovecii prophylaxis with trimethoprim-sulfamethoxazole (TMP-SMX) when neutropaenic.
- Iron chelation: Deferasirox (Jadenu® — PBS Authority Required) or deferoxamine if transfusional iron overload develops. Target ferritin <500 mcg/L.
- Oestrogen replacement: For female patients with primary ovarian insufficiency — physiological oestradiol with cyclic progesterone. Important for bone health, cardiovascular protection, and quality of life.
- Growth hormone: Recombinant GH (somatropin — PBS Authority Required) if documented GH deficiency and significant growth impairment.
- Psychosocial support: Referral to clinical psychology; peer support groups (Fanconi Anemia Research Fund — FARF); consideration of genetic counselling for the extended family.
- Pregnancy management: Pregnancy is possible in some female patients (especially after oestrogen replacement). High-risk obstetric care is recommended — increased risk of pre-eclampsia, preterm delivery, and small-for-gestational-age infants. Avoid teratogenic agents.
Special Populations
ATSI Health Considerations
📚 References
- 1. Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol. 2015;37:49–60.
- 2. Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer. 2018;18(3):168–185.
- 3. Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2007;2007:29–39.
- 4. Soulier J. Fanconi anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:492–497.
- 5. Ebens CL, MacMillan ML, Wagner JE. Hematopoietic cell transplantation in Fanconi anemia: current evidence and challenges. Hematol Oncol Clin North Am. 2018;32(4):713–727.
- 6. Peake JD, Noguchi E. Fanconi anemia: current insights regarding epidemiology, cancer, and DNA repair. Hum Genet. 2022;141(12):1841–1859.
- 7. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2023. Canberra: AIHW; 2023.
- 8. Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood. 2003;101(4):1249–1256.
- 9. Rosenberg PS, Tamary H, Alter BP. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi anemia in the United States and Israel. Am J Med Genet A. 2011;155A(8):1877–1883.
- 10. Mehta PA, Tolar J. Fanconi anemia. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington; 2024.
- 11. Australasian Bone Marrow Transplant Recipient Registry (ABMTRR). Annual Report 2023. Sydney: ABMTRR; 2023.
- 12. Savage SA, Dufour C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol. 2017;54(2):105–114.
- 13. Savage SA. Connecting Fanconi anemia with head and neck cancer. Clin Cancer Res. 2019;25(24):7258–7260.
- 14. Pagliuca S, Gurnari C, Hercus C, et al. The molecular landscape of Fanconi anemia: insights from genomics. Leukemia. 2023;37(5):991–1003.