📋 Key Information Summary
- Paroxysmal Nocturnal Haemoglobinuria (PNH) is a rare, acquired clonal haematopoietic stem cell disorder characterised by complement-mediated intravascular haemolysis, thrombophilia, and bone marrow failure.
- Caused by somatic mutations in the PIG-A gene leading to deficiency of GPI-anchored complement regulatory proteins CD55 and CD59 on affected blood cells.
- Classic triad: episodic intravascular haemolysis (dark urine, fatigue), cytopenias (often overlapping aplastic anaemia), and thrombosis (venous and arterial, including unusual sites such as hepatic veins).
- Thrombosis is the leading cause of death in untreated PNH and may occur in unusual sites — hepatic vein (Budd–Chiari), cerebral sinuses, mesenteric veins, dermal veins.
- Flow cytometry detecting CD55 and CD59 deficiency on red cells and granulocytes is the gold-standard diagnostic test; LDH is markedly elevated and the primary monitoring biomarker.
- Eculizumab (Soliris®) and ravulizumab (Ultomiris®) — terminal complement (C5) inhibitors — are the cornerstone of therapy for classic PNH with haemolysis or thrombosis; PBS Authority Required listing.
- Eculizumab/ravulizumab carry a significant risk of Neisseria meningitidis infection; meningococcal vaccination is mandatory ≥2 weeks before initiation, with ongoing prophylactic antibiotics.
- All patients require baseline meningococcal vaccination (ACWY and B strains) and indefinite antibiotic prophylaxis (penicillin V 500 mg BD or alternative) while on C5 inhibitor therapy.
- Anticoagulation is indicated for thrombotic events but primary thromboprophylaxis decisions are individualised; PNH-directed anticoagulation is not routinely recommended without C5 inhibitor therapy.
- Allogeneic haematopoietic stem cell transplantation (HSCT) is the only curative option, reserved for severe/refractory disease or severe aplastic anaemia component; transplant-related mortality remains significant.
- Pregnancy in PNH is high-risk (increased thrombosis and haemolysis); eculizumab can be continued in pregnancy and is preferred over ravulizumab due to more extensive safety data.
- Aboriginal and Torres Strait Islander patients face barriers including remote geographic access, limited specialist haematology services, and need for culturally safe care and regular monitoring pathways.
Introduction & Australian Epidemiology
Paroxysmal Nocturnal Haemoglobinuria (PNH) is a rare, acquired clonal haematopoietic stem cell disorder arising from somatic mutations in the PIG-A gene. These mutations impair biosynthesis of the glycosylphosphatidylinositol (GPI) anchor, rendering blood cells deficient in complement regulatory surface proteins — most critically CD55 (decay-accelerating factor) and CD59 (membrane inhibitor of reactive lysis). The result is complement-mediated intravascular haemolysis, a prothrombotic state, and variable degrees of bone marrow failure.
PNH is exceptionally rare, with an estimated prevalence of approximately 1–1.5 per 100,000 population in developed countries. In Australia, with a population of ~26 million, the total number of diagnosed PNH patients is estimated at 250–400 individuals, though underdiagnosis remains likely. The median age at diagnosis is 30–40 years, with a slight female predominance. PNH is frequently associated with, or evolves from, aplastic anaemia — approximately 30–50% of patients with aplastic anaemia harbour a detectable PNH clone.
The advent of complement-inhibitor therapy — first eculizumab (Soliris®, Alexion) and subsequently ravulizumab (Ultomiris®) — has transformed the natural history of PNH, reducing intravascular haemolysis, thrombotic risk, and transfusion dependence, and markedly improving survival. Access to these therapies in Australia is via the Pharmaceutical Benefits Scheme (PBS) Authority Required programme, managed through specialist haematology centres.
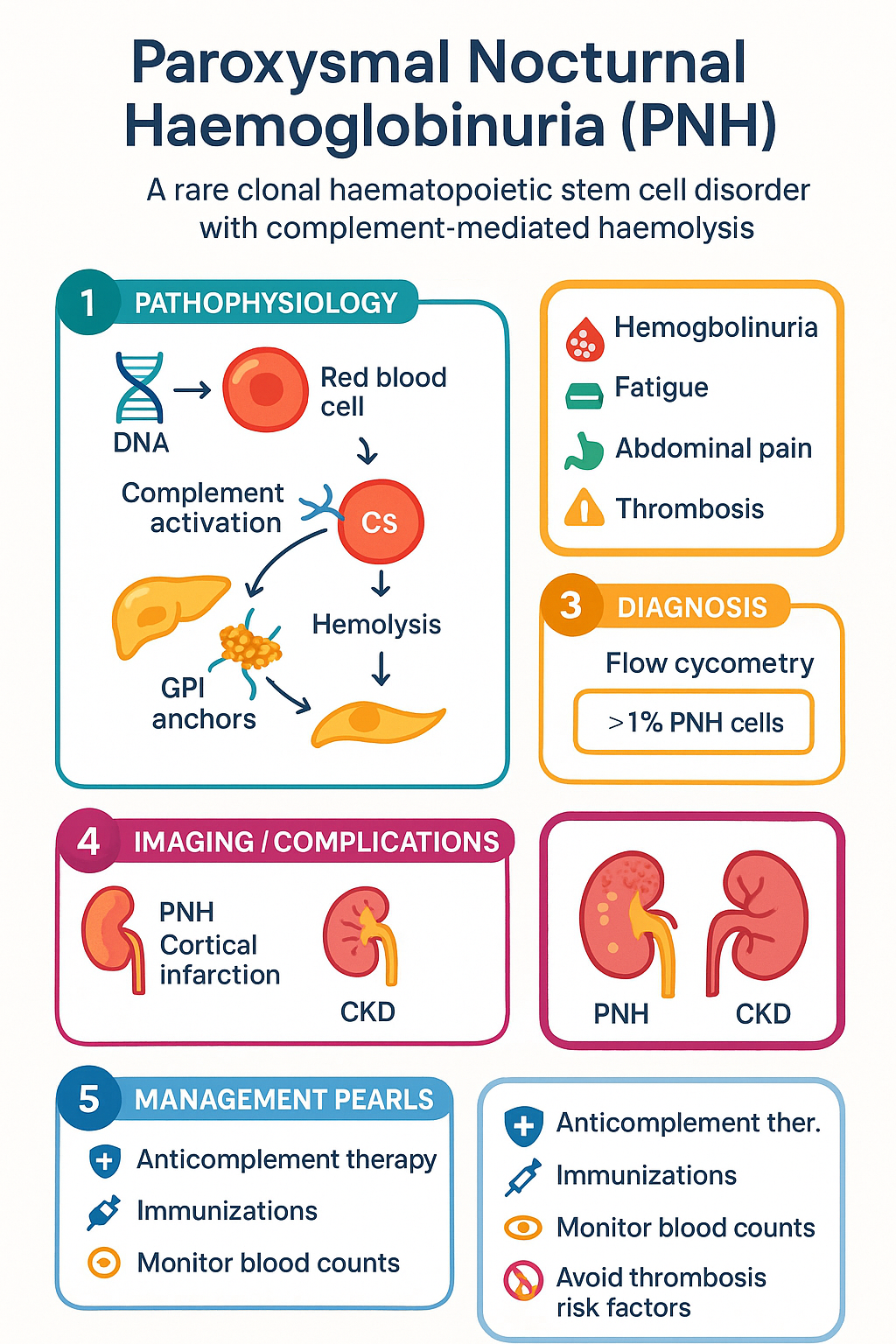
Pathogenesis — GPI Anchor Deficiency, CD55 & CD59
PNH arises from a somatic mutation in the X-linked PIG-A (phosphatidylinositol glycan class A) gene in a haematopoietic stem cell. Because the mutation is X-linked and males have only one copy, a single hit is sufficient; in females, random X-inactivation means that one hit in the active-X copy produces the phenotype. The mutation impairs the first step of GPI-anchor biosynthesis, preventing attachment of GPI-anchored proteins to the cell surface.
The GPI Anchor and Complement Regulation
The GPI anchor is a glycolipid structure that tethers numerous proteins to the extracellular surface of haematopoietic cells. Of particular relevance to PNH are two complement-regulatory proteins:
- CD55 (decay-accelerating factor, DAF): Accelerates decay of C3 and C5 convertases (C3bBb and C4b2a), limiting amplification of complement activation on the cell surface.
- CD59 (membrane inhibitor of reactive lysis, MIRL): Prevents incorporation of C9 into the C5b-9 membrane attack complex (MAC), directly blocking terminal complement-mediated lysis.
In PNH, the clonal expansion of PIG-A-mutated stem cells produces erythrocytes, granulocytes, monocytes, and platelets that lack both CD55 and CD59. This renders them exquisitely sensitive to complement activation. Under physiological conditions — triggered by infection, surgery, stress, or even the acidotic milieu of sleep (historically explaining the "nocturnal" component, though this mechanism is debated) — complement activation proceeds unimpeded, leading to direct intravascular haemolysis via MAC (C5b-9 deposition).
Clonal Dynamics
The PIG-A mutation alone is insufficient to explain the clonal dominance seen in PNH. Additional factors confer a selective growth advantage to the PNH clone, particularly in the setting of immune-mediated bone marrow suppression (overlapping aplastic anaemia). Hypothesised drivers include resistance of GPI-deficient stem cells to T-cell–mediated attack and altered cytokine signalling. Patients with large PNH clones (>50% GPI-deficient granulocytes) are more likely to develop clinically significant haemolysis and thrombosis.
Downstream Pathological Consequences
| Mechanism | Clinical Consequence |
|---|---|
| Complement-mediated intravascular haemolysis (C5b-9 MAC) | Anaemia, haemoglobinuria, fatigue, dysphagia, erectile dysfunction, abdominal pain |
| Free haemoglobin scavenges nitric oxide (NO) | Pulmonary hypertension, smooth muscle dystonia (dysphagia, abdominal pain, erectile dysfunction) |
| Platelet activation and NO depletion | Venous and arterial thrombosis — hepatic veins, cerebral sinuses, mesenteric, dermal |
| Chronic haemolysis and iron loss (urinary) | Iron deficiency (paradoxically, despite intravascular haemolysis), renal tubular injury |
| Underlying stem cell failure | Cytopenias (aplastic anaemia overlap), risk of MDS/AML transformation |
Clinical Features — Haemolysis, Thrombosis & Cytopenias
The clinical presentation of PNH is heterogeneous and often insidious. Many patients present with nonspecific symptoms for months to years before diagnosis. The disease is conventionally classified into three overlapping syndromes:
Other Clinical Features
- Smooth muscle dystonia: Oesophageal spasm (dysphagia), abdominal colic, erectile dysfunction — driven by NO depletion from free haemoglobin.
- Pulmonary hypertension: Due to chronic NO scavenging and possible thrombotic microangiopathy in pulmonary vasculature.
- Renal involvement: Chronic haemoglobinuria causes tubular iron deposition (haemosiderinuria), progressive renal impairment.
- Iron deficiency: Urinary iron loss through haemoglobinuria can be profound despite haemolytic anaemia — monitor ferritin and transferrin saturation.
- Severe paroxysms: Triggered by infection, surgery, stress, menstruation — may require transfusion support.
Investigations — Flow Cytometry & Biomarkers
Diagnosis of PNH requires a high index of clinical suspicion. The diagnostic pathway combines laboratory evidence of intravascular haemolysis with confirmation of GPI-anchor–deficient clones via flow cytometry.
Diagnostic Investigations
Imaging for Thrombosis
- CT pulmonary angiography (CTPA): Suspected pulmonary embolism.
- Doppler ultrasound: Hepatic vein (Budd–Chiari), portal vein, limb DVT.
- MRI venography: Cerebral venous sinus thrombosis.
- CT abdomen with contrast: Mesenteric vein thrombosis, hepatic vein thrombosis.
Management — Eculizumab, Anticoagulation & SCT
Management of PNH has been transformed by complement inhibition. Current Australian practice follows international consensus guidelines (International PNH Interest Group) adapted to the PBS and TGA regulatory framework.
Complement (C5) Inhibitor Therapy — The Cornerstone of Treatment
C5 inhibitors block terminal complement activation, preventing C5b-9 MAC formation and intravascular haemolysis. They are indicated for PNH patients with:
- Classic haemolytic PNH with elevated LDH (>1.5× ULN) and symptoms
- Thrombotic events attributable to PNH
- Transfusion-dependent anaemia with evidence of complement-mediated haemolysis
- Significant symptoms (fatigue, dysphagia, abdominal pain, erectile dysfunction) affecting quality of life
Anticoagulation
Thrombosis is the leading cause of morbidity and mortality in PNH. Anticoagulation management requires specialist haematology involvement.
| Scenario | Recommendation |
|---|---|
| Acute thrombosis | Immediate therapeutic anticoagulation (heparin → warfarin or DOAC) PLUS initiation of C5 inhibitor if not already receiving. Thrombolysis for life-threatening events (e.g., Budd–Chiari). |
| Prior thrombosis (on C5 inhibitor) | Long-term anticoagulation — usually warfarin (INR 2.0–3.0) or DOAC; specialist decision. Do NOT cease anticoagulation when starting eculizumab. |
| No prior thrombosis (on C5 inhibitor) | Primary thromboprophylaxis: individualised decision. C5 inhibitor therapy itself reduces thrombotic risk significantly. Low-dose aspirin may be considered but evidence is limited. DOAC/warfarin prophylaxis considered if high-risk features (large clone, prior history, pregnancy). |
| No C5 inhibitor therapy | Higher thrombotic risk. Consider anticoagulation prophylaxis in all patients with large PNH clones (>50% GPI-deficient granulocytes). Warfarin preferred over DOACs due to more limited evidence in PNH. |
Supportive Care
- Iron supplementation: Treat iron deficiency aggressively (IV iron preferred if severe, oral iron may exacerbate haemolysis in some patients). Target ferritin >50 µg/L, transferrin saturation >20%.
- Folic acid: 5 mg PO daily (increased folate demand from chronic haemolysis).
- Folic acid: 5 mg PO daily to support erythropoiesis.
- Blood transfusion: Leucodepleted, CMV-safe red cells. Avoid unnecessary transfusions to minimise alloimmunisation risk.
- EPO: Consider if anaemia persists despite C5 inhibitor and iron repletion (limited evidence).
- Renal monitoring: Regular eGFR monitoring; manage chronic kidney disease per standard guidelines.
- Pulmonary hypertension screening: Echocardiography at baseline and annually if symptoms (dyspnoea) develop.
Allogeneic Haematopoietic Stem Cell Transplantation (HSCT)
Allogeneic HSCT is the only curative therapy for PNH, replacing the defective haematopoietic stem cell clone. However, transplant-related morbidity and mortality (10–30% depending on conditioning and donor type) limit its role. In the era of C5 inhibitors, HSCT is reserved for:
- PNH with severe aplastic anaemia component refractory to immunosuppressive therapy
- C5 inhibitor–refractory disease (breakthrough haemolysis despite adequate complement inhibition, or evolving to aplastic anaemia / MDS)
- Life-threatening thrombotic events despite adequate C5 inhibitor therapy and anticoagulation
- Young patients with matched sibling donors and high-risk features (specialist decision)
Transplant should be performed at experienced haematopoietic stem cell transplant centres (e.g., Royal Adelaide Hospital, Westmead Hospital, Peter MacCallum Cancer Centre). Consultation with a transplant physician is essential for all patients at diagnosis for risk stratification and future planning.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
PNH is rare and there are no population-specific prevalence data for Aboriginal and Torres Strait Islander Australians. However, the high burden of comorbid chronic disease, barriers to specialist access, and unique cultural considerations require specific attention when caring for Indigenous patients with PNH.
Monitoring
Ongoing monitoring of PNH patients on C5 inhibitor therapy requires a structured approach, ideally coordinated through a specialist haematology centre.
📚 References
- 1. Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nature Reviews Disease Primers. 2017;3:17028.
- 2. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–3709.
- 3. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804–2811.
- 4. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. New England Journal of Medicine. 2006;355(12):1233–1243.
- 5. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539.
- 6. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor–experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549.
- 7. Socié G, Caby-Tosi MP, Marantz JL, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and pregnancy. European Journal of Haematology. 2019;102(2):150–158.
- 8. De Latour RP, Mary JY, Salanoubat C, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8):3099–3106.
- 9. Australian Technical Advisory Group on Immunisation (ATAGI). Australian Immunisation Handbook. Australian Government Department of Health; 2024. Meningococcal disease chapter.
- 10. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework. Canberra: AIHW; 2023.
- 11. Richards SJ, Hill A, Hillmen P. Recent advances in the diagnosis, monitoring, and management of patients with paroxysmal nocturnal hemoglobinuria. Cytometry Part B: Clinical Cytometry. 2007;72B(5):291–298.
- 12. Röth A, Rottinghaus ST, Hill A, et al. Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of two phase 3 studies (301/302). Blood. 2019;134(Supplement_1):2043.