📋 Key Information Summary
- Myelodysplastic syndromes (MDS) are clonal haematopoietic stem cell disorders characterised by ineffective haematopoiesis, peripheral blood cytopenias, dysplastic morphology, and risk of transformation to acute myeloid leukaemia (AML).
- Median age at diagnosis is approximately 72 years; incidence in Australia is ~3–5 per 100,000 per year, rising to >20 per 100,000 in patients aged >80 years.
- IPSS-R (Revised International Prognostic Scoring System) is the standard risk-stratification tool, incorporating cytogenetics, blast percentage, haemoglobin, platelet count, and absolute neutrophil count to classify patients into five risk groups (Very Low to Very High).
- Key cytogenetic abnormalities include del(5q), −7/del(7q), trisomy 8, del(20q), and complex karyotype (≥3 abnormalities), which directly influence IPSS-R score and therapeutic decisions.
- Somatic mutations in genes such as SF3B1, TP53, ASXL1, RUNX1, DNMT3A, and TET2 are increasingly used to refine prognosis and guide therapy; TP53 mutations confer particularly poor outcomes.
- Supportive care with transfusions and erythropoiesis-stimulating agents (ESAs) is appropriate for lower-risk MDS, while hypomethylating agents (azacitidine) are standard for higher-risk disease.
- Azacitidine (Vidaza®) is PBS-listed and remains the first-line agent for higher-risk MDS; it has demonstrated a median overall survival benefit of ~9.3 months compared with conventional care regimens.
- Allogeneic stem cell transplantation (allo-SCT) remains the only potentially curative treatment, considered for fit patients aged typically ≤70 years with intermediate-2 or high-risk disease.
- Lenalidomide (Revlimid®) is the preferred treatment for lower-risk MDS with isolated del(5q), achieving transfusion independence in approximately 67% of patients.
- Aboriginal and Torres Strait Islander patients may present with more advanced disease due to barriers in accessing specialist haematology services and diagnostic bone marrow biopsies, particularly in remote communities.
- Iron overload from chronic transfusion support can cause cardiac, hepatic, and endocrine toxicity; serum ferritin should be monitored and iron chelation therapy (deferasirox) considered when ferritin >1,000 µg/L with ongoing transfusion need.
- All patients should be referred to a multidisciplinary haematology team for risk stratification and treatment planning; clinical trial participation should be encouraged where available.
Introduction & Australian Epidemiology
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal haematopoietic stem cell disorders characterised by ineffective haematopoiesis, peripheral blood cytopenias, and morphological dysplasia in one or more cell lineages. The disease carries a variable risk of progression to acute myeloid leukaemia (AML), ranging from <5% in low-risk subtypes to >50% in high-risk subtypes.
In Australia, MDS is predominantly a disease of the elderly, with a median age at diagnosis of approximately 72 years. The overall annual incidence is estimated at 3–5 per 100,000 population, though this rises sharply with age, exceeding 20 per 100,000 in patients over 80 years. Australian population-based data from the AIHW and cancer registries suggest that MDS is under-reported, as many lower-risk cases are diagnosed in the community without formal bone marrow examination.
De novo (primary) MDS accounts for approximately 80–85% of cases; the remainder are therapy-related MDS (t-MDS), arising after prior cytotoxic chemotherapy (particularly alkylating agents and topoisomerase II inhibitors) or radiation exposure. Secondary MDS is typically associated with complex cytogenetics and poorer prognosis.
Advances in next-generation sequencing (NGS) have revealed that >90% of MDS patients harbour at least one somatic mutation, with a median of 2–4 driver mutations per patient. This molecular understanding has been incorporated into the WHO 2022 and ICC 2022 classification systems, which integrate genetic data alongside morphology and cytogenetics.
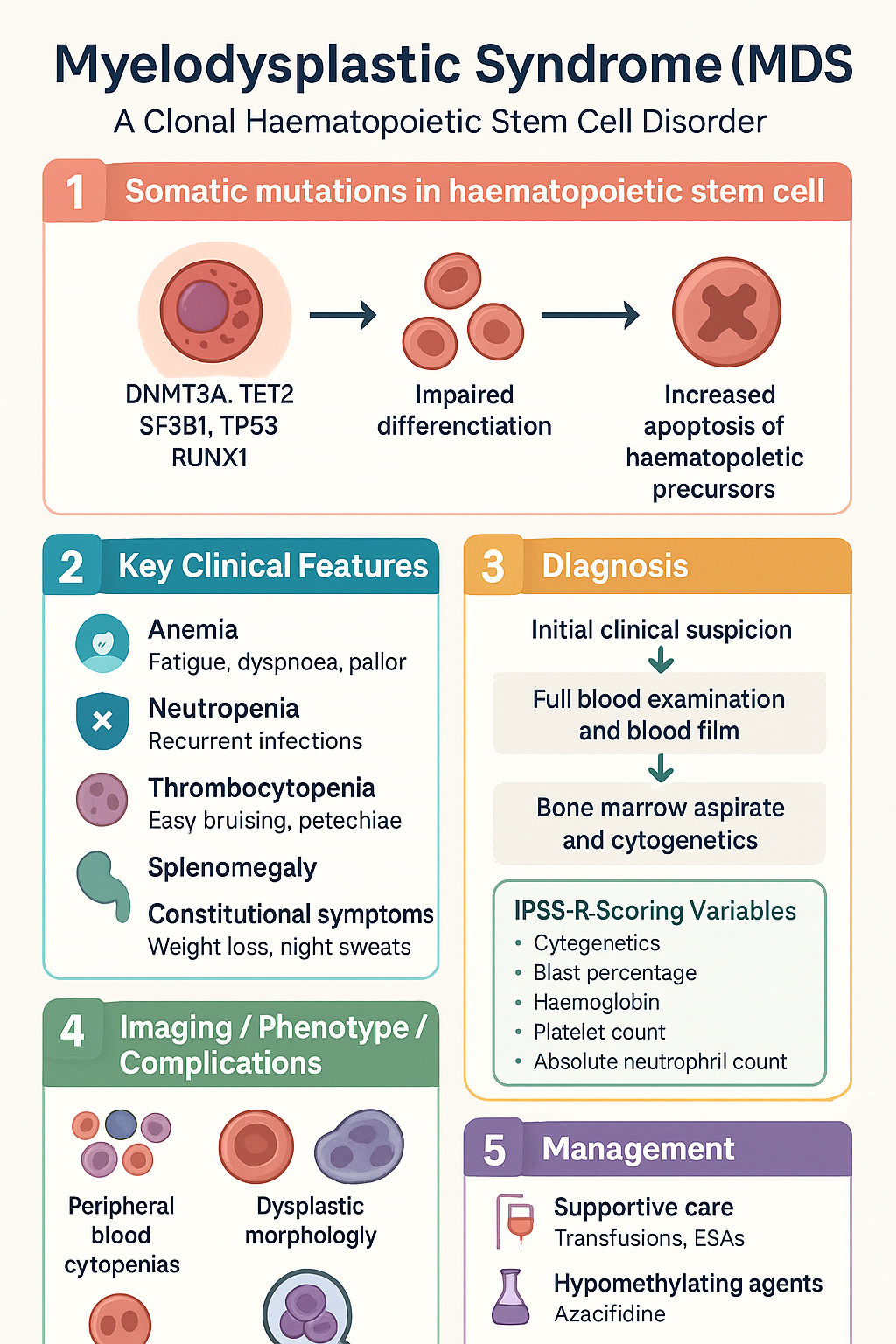
Pathogenesis & Cytogenetics
Pathophysiology
MDS arises from the acquisition of somatic mutations in a haematopoietic stem cell, leading to clonal expansion with impaired differentiation and increased apoptosis of haematopoietic precursors. The paradox of peripheral cytopenias despite a hypercellular bone marrow reflects ineffective haematopoiesis — cells are produced but are destroyed prematurely within the marrow or are functionally defective.
Several non-mutually exclusive mechanisms contribute:
- Intrinsic stem cell defect: Somatic mutations in epigenetic regulators (DNMT3A, TET2, IDH1/2), RNA splicing factors (SF3B1, SRSF2, U2AF1, ZRSR2), transcription factors (RUNX1, ETV6), cohesin complex, and signal transduction pathways (JAK2, FLT3).
- Clonal haematopoiesis of indeterminate potential (CHIP): Age-related clonal mutations (particularly DNMT3A, TET2, ASXL1) are found in ~10% of individuals aged >70 without cytopenias; ~0.5–1% per year progress to overt MDS or AML.
- Bone marrow microenvironment dysfunction: Abnormal stromal signalling, immune dysregulation (including T-cell mediated suppression of normal haematopoiesis), and altered cytokine milieu contribute to cytopenias.
- Epigenetic silencing: Aberrant DNA methylation and histone modification silence tumour suppressor genes and differentiation pathways, forming the rationale for hypomethylating agent therapy.
Cytogenetic Abnormalities
Conventional cytogenetics (G-banded karyotyping) is mandatory at diagnosis and is a core component of IPSS-R scoring. Approximately 40–50% of de novo MDS patients have a detectable abnormality at presentation.
| Cytogenetic Abnormality | Frequency | IPSS-R Cytogenetic Group | Clinical Significance |
|---|---|---|---|
| Normal karyotype | ~50% | Good | Favourable prognosis; often associated with SF3B1 mutation and ring sideroblasts |
| del(5q) alone | ~10–15% | Good | Defines 5q- syndrome (typical in older women); excellent response to lenalidomide |
| −Y, del(11q), del(12p), del(20q) | ~5–10% | Good | Generally indolent course |
| Trisomy 8 | ~5–10% | Intermediate | May benefit from immunosuppressive therapy in selected cases |
| −7/del(7q) | ~5–10% | Poor | Aggressive; associated with prior chemotherapy or environmental exposures |
| Complex karyotype (≥3 abnormalities) | ~10–15% | Very Poor | High AML transformation risk; often TP53 mutated; consider SCT early |
| i(17q), inv(3)/t(3q) | Rare | Very Poor | Extremely poor prognosis; often refractory to standard therapy |
Key Somatic Mutations in MDS
Next-generation sequencing (NGS) myeloid panels are increasingly recommended at diagnosis, particularly for refining prognosis in intermediate-risk IPSS-R and for guiding therapeutic decisions.
| Gene/Pathway | Frequency in MDS | Prognostic Impact |
|---|---|---|
| SF3B1 (splicing) | ~25–30% | Favourable; associated with ring sideroblasts, lower AML risk |
| TET2 (epigenetic) | ~20–25% | Variable; may predict response to azacitidine |
| ASXL1 (chromatin) | ~15–20% | Unfavourable; independent adverse prognostic factor |
| DNMT3A (epigenetic) | ~10–15% | Unfavourable; common in CHIP → MDS progression |
| TP53 (tumour suppressor) | ~5–10% | Very unfavourable; associated with complex karyotype, therapy-related MDS, poor SCT outcomes |
| RUNX1 (transcription factor) | ~8–10% | Unfavourable; increased AML transformation risk |
| SRSF2 (splicing) | ~10–15% | Intermediate–unfavourable; common in CMML overlap |
| IDH1/IDH2 (metabolic) | ~5–10% | Variable; potential target for IDH inhibitors |
Classification (IPSS-R Scoring)
WHO 2022 Classification of MDS
The WHO 2022 classification has reorganised MDS subtypes to incorporate molecular genetic data. Key changes include the recognition of MDS with mutated SF3B1 as a distinct entity (≥5% ring sideroblasts + SF3B1 mutation) and the creation of MDS with biallelic TP53 inactivation as a separate high-risk category.
| WHO 2022 Subtype | Key Features | Median Survival |
|---|---|---|
| MDS with SF3B1 mutation | ≥5% ring sideroblasts + SF3B1 mutation; single lineage dysplasia | >5 years |
| MDS with del(5q) | Isolated del(5q); megakaryocyte hypolobation; usually anaemia only | >5 years with lenalidomide |
| MDS, low blast count (MDS-LB) | <5% marrow blasts; 1–2 lineage dysplasia; <1 × 10⁹/L blasts | 3–5 years |
| MDS, high blast count (MDS-HB) | 5–9% marrow blasts (MDS-HB1) or 10–19% (MDS-HB2) | 1–2 years |
| MDS with fibrosis | Moderate–severe reticulin fibrosis (MF-2/3); cytopenias; splenomegaly | ~2 years |
| MDS/AML | 20–29% marrow blasts; biologically closer to AML | <1 year without AML therapy |
| MDS with biallelic TP53 inactivation | Two TP53 hits (mutation + deletion); complex karyotype common | ~6–12 months |
Revised International Prognostic Scoring System (IPSS-R)
The IPSS-R remains the most widely validated prognostic tool for MDS. It integrates five variables to assign a score from 0 to 10, categorising patients into five risk groups. It should be calculated at diagnosis and reassessed at disease milestones.
IPSS-R Scoring Variables
| Prognostic Variable | Score 0 | Score 0.5 | Score 1 | Score 1.5 | Score 2 | Score 3 | Score 4 |
|---|---|---|---|---|---|---|---|
| Cytogenetics | Very Good | — | Good | Intermediate | Poor | Very Poor | — |
| BM blasts (%) | ≤2% | — | >2–<5% | 5–10% | >10% | — | — |
| Hb (g/L) | ≥100 | — | 80–<100 | <80 | — | — | — |
| Platelets (×10⁹/L) | ≥100 | 50–<100 | <50 | — | — | — | — |
| ANC (×10⁹/L) | ≥0.8 | <0.8 | — | — | — | — | — |
IPSS-R Risk Categories
Clinical Features & Blood Film
Clinical Presentation
MDS is frequently asymptomatic at presentation, discovered incidentally on a routine full blood examination (FBE) showing cytopenias. When symptomatic, features reflect the specific cytopenias:
- Anaemia (most common, ~85%): Fatigue, dyspnoea on exertion, pallor, palpitations, dizziness. Often macrocytic or normocytic.
- Neutropenia (~30–50%): Recurrent or unusual infections, oral ulceration, fever. Severe neutropenia (ANC <0.5 × 10⁹/L) increases infection risk.
- Thrombocytopenia (~20–40%): Easy bruising, petechiae, epistaxis, gum bleeding, menorrhagia. Platelet function may also be impaired despite normal counts.
- Splenomegaly: Present in ~10–20%, more common in chronic myelomonocytic leukaemia (CMML) and MDS/myeloproliferative neoplasm (MPN) overlap syndromes.
- Constitutional symptoms: Unexplained weight loss, night sweats, and fever may indicate transformation or concomitant infection.
- Skin manifestations: Sweet syndrome (acute febrile neutrophilic dermatosis) or leukaemia cutis can rarely accompany MDS.
Blood Film Findings
A peripheral blood film should be reviewed by a haematologist in all suspected cases. Characteristic findings include:
- Red cells: Macro-ovalocytes (oval macrocytes), anisocytosis, poikilocytosis, basophilic stippling. Ring sideroblasts are not visible on blood film but cause a dimorphic red cell population.
- Neutrophils: Hypogranular cytoplasm, pseudo–Pelger–Huët anomaly (bilobed nuclei), nuclear abnormalities (ring nuclei, hypersegmentation).
- Platelets: Hypogranular or agranular platelets, giant platelets, micromegakaryocyte fragments.
- Blasts: Circulating blasts may be present; even 1% circulating blasts has prognostic significance and warrants bone marrow evaluation.
Investigations at Diagnosis
Management
Management of MDS is guided by IPSS-R risk category, patient fitness (comorbidities, performance status, age), and treatment goals. The overarching objectives are to improve quality of life, reduce transfusion dependence, delay disease progression, and, where possible, extend survival or achieve cure.
1. Supportive Care (All Risk Categories)
Supportive care is the cornerstone of management for lower-risk MDS and an essential adjunct for higher-risk disease.
Transfusion Support
- Red cell transfusions: Maintain Hb ≥80–100 g/L (individualised based on symptoms). Use irradiated blood products for all MDS patients (reduced risk of transfusion-associated graft-versus-host disease, especially if SCT is considered). Leucodepleted products are standard in Australia.
- Platelet transfusions: For active bleeding with platelets <30 × 10⁹/L, or prophylactically <10 × 10⁹/L. Refractoriness may develop with repeated transfusions — assess anti-HLA antibodies.
- Iron chelation therapy: Consider when serum ferritin >1,000 µg/L with ongoing transfusion need. Deferasirox (Jadenu®/Desferrioxamine) is the preferred oral agent; PBS Authority Required for MDS-related iron overload.
Erythropoiesis-Stimulating Agents (ESAs)
ESAs are recommended for lower-risk MDS (IPSS-R Very Low, Low, Intermediate) with symptomatic anaemia and serum EPO <200 U/L. Response rates are approximately 40–60% in this group.
Infection Prophylaxis
- Antimicrobial prophylaxis: Consider fluoroquinolone prophylaxis (ciprofloxacin 500 mg PO BD) when ANC <0.5 × 10⁹/L. Antifungal prophylaxis (fluconazole or posaconazole) for prolonged neutropenia. Antiviral prophylaxis (aciclovir) if prior HSV/VZV infection.
- Vaccinations: Annual influenza, pneumococcal (Prevenar 13 + Pneumovax 23), COVID-19, and Zostavax/Shingrix (recombinant preferred in immunocompromised) as per ATAGI guidelines.
2. Lower-Risk MDS: Disease-Modifying Therapies
Lenalidomide for del(5q) Syndrome
Lenalidomide is the treatment of choice for lower-risk MDS with isolated del(5q). It achieves transfusion independence in ~67% of patients, with durable responses (median duration >2 years) and cytogenetic remissions in ~45%.
Luspatercept for Ring Sideroblast MDS
Luspatercept (Reblozyl®) is a first-in-class erythroid maturation agent approved for lower-risk MDS with ring sideroblasts (with or without SF3B1 mutation) who have failed or are ineligible for ESAs. It is PBS-listed in Australia.
3. Higher-Risk MDS: Hypomethylating Agents
Azacitidine is the standard first-line therapy for higher-risk MDS (IPSS-R Intermediate, High, Very High) and remains the most widely used hypomethylating agent in Australia.
Alternatives to Azacitidine
- Decitabine (Dacogen®): Not PBS-listed for MDS in Australia. 20 mg/m² IV daily for 5 days every 28 days. Similar efficacy to azacitidine; used if azacitidine intolerance.
- Venetoclax + azacitidine: Off-label in MDS but increasingly used in MDS-AML overlap (20–29% blasts) based on extrapolation from AML data. PBS-listed for AML only.
4. Allogeneic Stem Cell Transplantation (allo-SCT)
Allogeneic SCT remains the only potentially curative treatment for MDS. Eligibility is determined by patient fitness, age, comorbidities, IPSS-R risk, donor availability, and patient preference.
| Factor | Consideration |
|---|---|
| Age | Typically ≤70 years for myeloablative; reduced-intensity conditioning (RIC) may extend to 75 |
| IPSS-R | Strongest indication in High and Very High risk. Intermediate risk: consider if adverse molecular features (TP53, RUNX1) or transfusion-dependent |
| Donor | Matched sibling donor (MSD) preferred; MUD acceptable. Haploidentical and cord blood as alternatives |
| Timing | Refer early for workup. Bridging with azacitidine is common while donor search proceeds. Delay >6 months may worsen outcomes |
| Comorbidities | HCT-CI (Haematopoietic Cell Transplantation-Comorbidity Index) to assess fitness |
| TP53 mutation | Poor SCT outcomes overall, but still considered the best option. Novel approaches (post-SCT maintenance with azacitidine ± venetoclax) under investigation |
5. Experimental and Emerging Therapies
- Imetelstat: Telomerase inhibitor with promising results in lower-risk, transfusion-dependent MDS after ESA failure. FDA-approved (2024); PBS listing pending.
- Magtacimab (anti-TIM-3): Investigational immunotherapy under clinical trial evaluation in higher-risk MDS.
- IDH inhibitors (ivosidenib, enasidenib): Targeted therapy for IDH1/IDH2-mutated MDS/AML. Limited PBS access; consider via Special Access Scheme.
- Venetoclax combinations: Venetoclax + azacitidine under investigation in higher-risk MDS. Extrapolation from AML data; not yet standard of care in MDS.
- Clinical trials: Patients should be considered for clinical trial participation where available. Australian trial registries: ANZCTR, Australian Cancer Trials, and site-specific haematology trials.
Special Populations
Pregnancy
Paediatric MDS
Elderly Patients (>75 years)
Renal Impairment
Hepatic Impairment
Immunocompromised / Post-Transplant
Aboriginal and Torres Strait Islander Health Considerations
Monitoring & Follow-Up
Ongoing monitoring is essential for all MDS patients regardless of treatment pathway. The frequency of follow-up should be tailored to the IPSS-R risk category and the treatment being administered.
📚 References
- 1. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465.
- 2. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703–1719.
- 3. Bernard E, Tuechler H, Greenberg PL, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evidence. 2022;1(7):EVIDoa2200008.
- 4. Fenaux P, Mufti GJ, Hellström-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232.
- 5. List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456–1465.
- 6. Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. 2020;382(2):140–151.
- 7. Australasian Leukaemia & Lymphoma Group. Australian guidelines for the management of myelodysplastic syndromes. ALLG; 2023.
- 8. Australian Institute of Health and Welfare. Cancer in Australia 2024. Canberra: AIHW; 2024.
- 9. Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943–2964.
- 10. Sekeres MA, Guyatt G, Abel G, et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020;4(15):3528–3549.
- 11. National Health and Medical Research Council. Clinical practice guidelines for the management of myelodysplastic syndromes. NHMRC; 2022.
- 12. Valent P, Horny HP, Bennett JM, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: consensus statements and report from a working conference. Leuk Res. 2007;31(6):727–736.
- 13. de Witte T, Bowen D, Robin M, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood. 2017;129(13):1753–1762.
- 14. Platzbecker U, Santini V, Fenaux P, et al. Imetelstat in patients with lower-risk myelodysplastic syndromes who have relapsed or are refractory to erythropoiesis-stimulating agents (IMerge): results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet. 2024;403(10423):231–242.
- 15. RACGP. RACGP Red Book: guidelines for preventive activities in general practice. 10th ed. Melbourne: RACGP; 2024.