📋 Key Information Summary
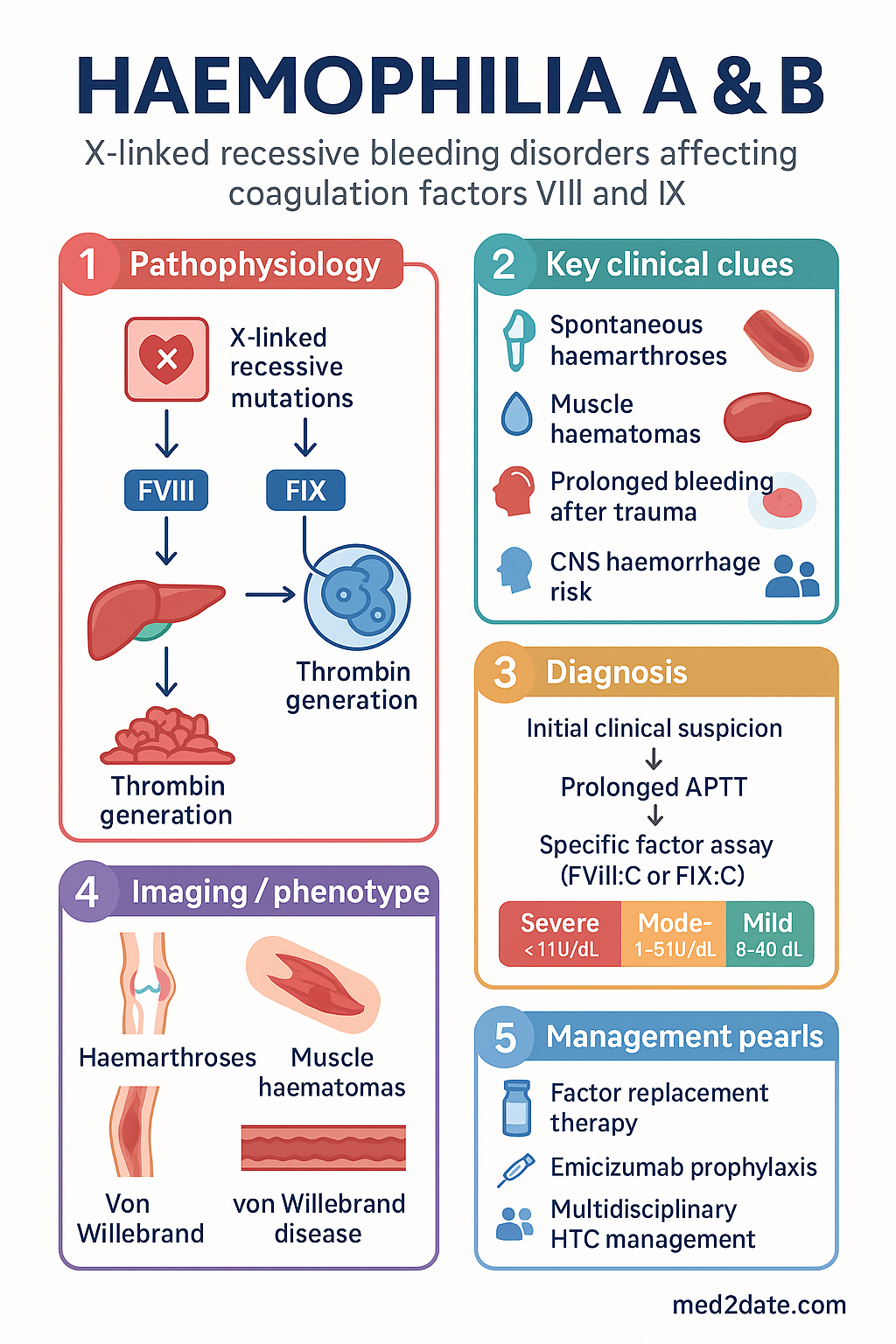
- Haemophilia A (factor VIII deficiency) and B (factor IX deficiency) are X-linked recessive bleeding disorders predominantly affecting males.
- Severity is classified by baseline clotting factor level: Severe (<1 IU/dL), Moderate (1–5 IU/dL), Mild (>5–40 IU/dL).
- Severe haemophilia presents with spontaneous haemarthroses and muscle haematomas, typically from 6–18 months of age.
- Diagnosis relies on a prolonged APTT with normal PT; factor-specific assays confirm type and severity.
- Inhibitor development (alloantibodies) is the major treatment complication, occurring in ~30% of severe Haemophilia A.
- Management centres on factor replacement therapy, with the goal of prophylaxis to prevent bleeding and arthropathy.
- Emicizumab (Hemlibra®), a subcutaneous bispecific monoclonal antibody, is the standard prophylaxis for Haemophilia A (± inhibitors).
- On-demand treatment for acute bleeding requires prompt factor concentrate infusion to achieve haemostatic levels.
- Patients must be managed in a multidisciplinary Haemophilia Treatment Centre (HTC) and carry an emergency treatment plan.
- Mandatory physiotherapy and joint health monitoring are integral to prevent chronic arthropathy.
- Live vaccines are generally contraindicated in neonates on high-dose prophylactic factor due to theoretical risk of vaccine-strain bleeding.
- Genetic counselling and carrier testing are essential for affected families.
- Aboriginal and Torres Strait Islander communities face significant barriers to accessing specialised haemophilia care.
Introduction & Australian Epidemiology
Haemophilia A (factor VIII [FVIII] deficiency) and Haemophilia B (factor IX [FIX] deficiency, also known as Christmas disease) are congenital, X-linked recessive bleeding disorders. They primarily affect males, with females usually being asymptomatic carriers. The conditions are characterised by deficient or defective coagulation factor proteins, leading to a spectrum of bleeding manifestations ranging from spontaneous haemarthroses and deep muscle haematomas to life-threatening intracranial or surgical haemorrhage.
In Australia, haemophilia is classified as a rare disease. The Australian Bleeding Disorders Registry (ABDR) provides key epidemiological data. Haemophilia A has an incidence of approximately 1 in 10,000 male births, while Haemophilia B is less common at approximately 1 in 50,000 male births. Nationally, there are over 3,000 diagnosed patients. The prevalence among Aboriginal and Torres Strait Islander peoples is not well characterised due to under-diagnosis and access barriers, but is estimated to be similar to the non-Indigenous population. Management is coordinated through a national network of specialised Haemophilia Treatment Centres (HTCs).
Genetics & Factor Deficiencies
Inheritance & Molecular Basis
Both haemophilia A and B follow an X-linked recessive inheritance pattern. The FVIII gene (F8) is located on the long arm of the X chromosome (Xq28). Common mutations in severe haemophilia A include intron 22 inversions (~45% of severe cases) and large deletions. The FIX gene (F9) is on Xq27.1; a wider variety of missense mutations are seen in haemophilia B. A negative family history is present in ~30% of cases due to de novo mutations.
Factor Deficiencies
FVIII and FIX are components of the intrinsic tenase complex. FVIII circulates bound to von Willebrand factor (vWF), which protects it from premature clearance. FIX is a vitamin K-dependent serine protease synthesised in the liver. A deficiency in either factor results in impaired thrombin generation, which is necessary for stable fibrin clot formation.
Inhibitors
Inhibitors are neutralising IgG alloantibodies that develop against the infused therapeutic factor. They are the most significant treatment complication. Risk factors include: severe deficiency, specific F8 gene mutations (e.g., large deletions, intron 22 inversion), family history of inhibitors, intensive first exposure to factor (e.g., surgical prophylaxis), and younger age at first treatment. Inhibitors are classified as low-titre (<5 Bethesda Units [BU]) or high-titre (≥5 BU).
Clinical Features & Severity Classification
Bleeding Phenotypes by Severity
Common Bleeding Manifestations
- Haemarthroses: Hallmark of severe disease. Most commonly affects knees, elbows, and ankles. Acute episodes present with pain, swelling, warmth, and reduced range of motion.
- Muscle/Soft Tissue Haematomas: Iliopsoas bleed (mimics appendicitis), calf, and forearm bleeds. Risk of compartment syndrome.
- CNS Haemorrhage: Leading cause of haemophilia-related death. Intracranial (especially post-traumatic) and extracranial (subgaleal) in neonates.
- Oral/Mucosal Bleeding: Prolonged bleeding after dental work, tongue/lip lacerations.
- Haematuria: Often spontaneous, requires urological investigation to exclude structural cause.
Investigations
Initial Screening & Diagnostic Tests
Genetic Testing
Available through specialised genetics services (MBS Item 73286). Indicated for definitive diagnosis, carrier testing for female relatives, and prenatal counselling. Identifies the specific mutation, which has prognostic value for inhibitor risk.
Management
1. Factor Replacement Therapy
The cornerstone of haemophilia management. In Australia, factor concentrates are provided free-of-charge to eligible patients through the national Product & Stewardship Programme, managed by the National Blood Authority (NBA).
2. Emicizumab (Hemlibra®) – For Haemophilia A
A subcutaneous, bispecific monoclonal antibody that mimics the cofactor function of FVIIIa by bridging FIXa and FX. It is the standard of care for prophylaxis in severe Haemophilia A, regardless of inhibitor status.
3. Management of Inhibitors
For patients with inhibitors, bypassing agents are used for treatment and prevention of bleeding.
4. Adjunctive & Supportive Therapies
- Analgesia: Paracetamol (avoid NSAIDs/COX-2 inhibitors due to platelet inhibition; opioids for severe pain).
- Physiotherapy: Essential for joint rehabilitation and musculoskeletal health. Regular assessment with the Haemophilia Joint Health Score (HJHS).
- RICE: Rest, Ice, Compression, Elevation as first aid for acute soft tissue/joint bleeds.
- Desmopressin (DDAVP): For mild haemophilia A only (stimulates release of stored FVIII). Test dose required. 0.3 µg/kg SC/IV or intranasal spray (Octim®).
Monitoring
Comprehensive, lifelong monitoring is managed by the HTC. Key components include:
- Bleeding Diaries: Patients record all bleeding episodes (location, cause, treatment).
- Annual Review: Comprehensive assessment including physical exam, joint health (HJHS), inhibitor screening (annually for first 50 exposure days, then every 3–5 years if low-titre), musculoskeletal ultrasound/MRI, quality of life, and psychosocial evaluation.
- Pharmacokinetic (PK)-Guided Dosing: Used to optimise prophylactic regimens based on individual factor half-lives.
- Inhibitor Surveillance: Mandatory testing before any planned surgery, after intensive treatment episodes, and if clinical response to factor is suboptimal.
- Imaging: Regular MRI (preferred) or ultrasound of index joints to detect early synovitis or cartilage damage (chronic arthropathy).
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. National Blood Authority Australia. National Haemophilia Management Guidelines. Canberra: NBA; 2023.
- 2. Australian Haemophilia Centre Directors' Organisation (AHCDO). Australian Bleeding Disorders Registry (ABDR) Annual Report 2022-2023. AHCDO; 2023.
- 3. Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158.
- 4. Collins PW, Fischer K, Morfini M, et al. Implications of coagulation factor VIII and IX pharmacokinetics for the clinical management of haemophilia. Haemophilia. 2021;27(1):4-17.
- 5. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N Engl J Med. 2017;377(9):809-818.
- 6. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935-1939.
- 7. Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice. 9th edn. East Melbourne: RACGP; 2016. (Genetic counselling section).
- 8. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2020 summary report. Canberra: AIHW; 2020.
- 9. Feldman BM, Funk SM, Bergstrom BM, et al. Validation of a new pediatric joint scoring tool from the International Hemophilia Prophylaxis Study Group: validity of the hemophilia joint health score (HJHS). Arthritis Care Res. 2011;63(2):223-230.
- 10. Castaman G, Linari S. Diagnosis and Treatment of Acquired Hemophilia A and B: Current Standards and Emerging Therapies. J Clin Med. 2021;10(16):3580.