📋 Key Information Summary
- Von Willebrand disease (VWD) is the most common inherited bleeding disorder, affecting approximately 1% of the general population, though clinically significant disease affects ~0.01%.
- Type 1 VWD (partial quantitative deficiency of VWF) accounts for ~70–80% of cases; Type 2 (qualitative defects) ~15–20%; Type 3 (virtual absence of VWF) is rare (<5%).
- Type 1 is autosomal dominant with variable penetrance; Types 2A, 2B, 2M are usually autosomal dominant; Type 2N and Type 3 are autosomal recessive.
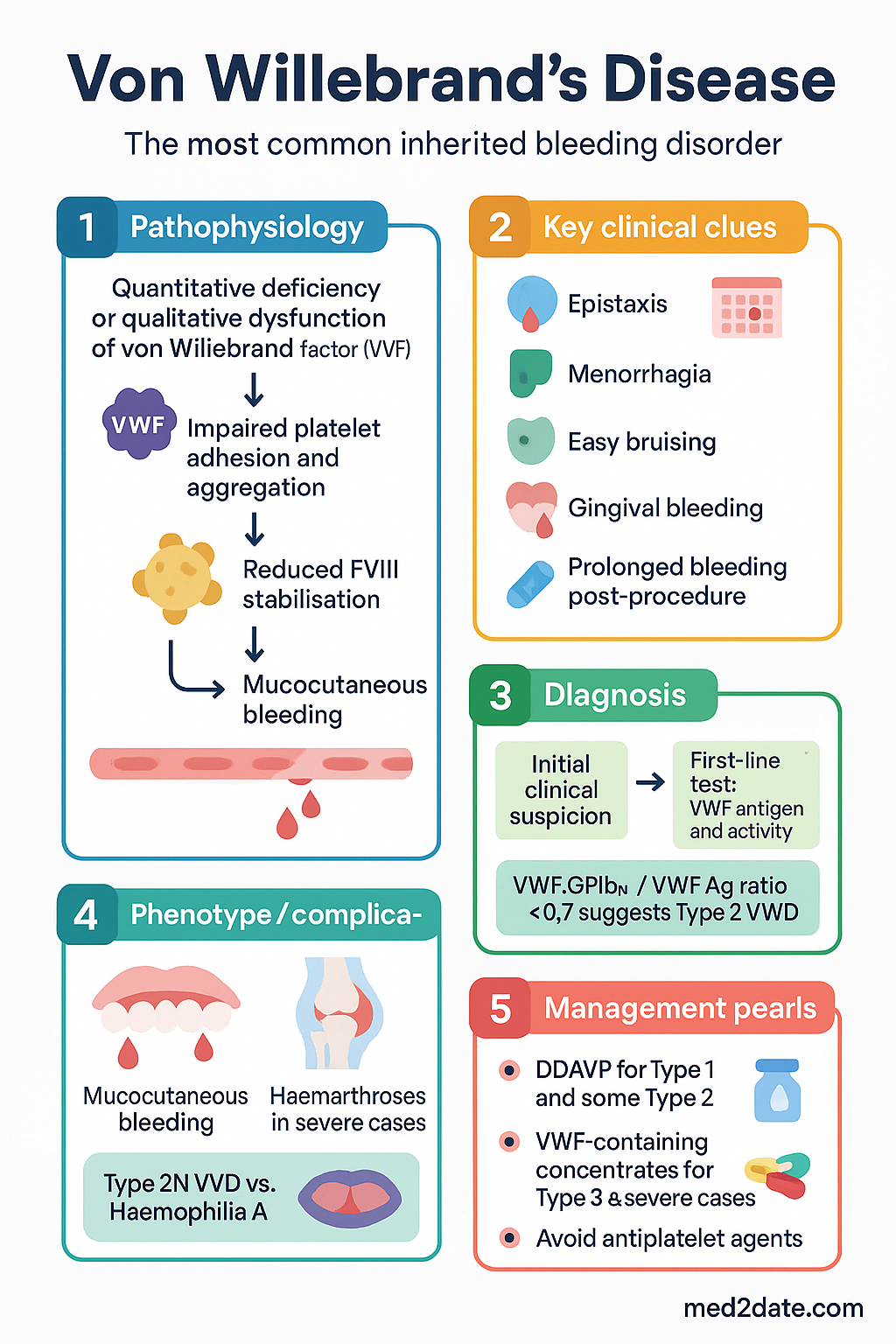
- The hallmark clinical presentation is mucocutaneous bleeding — epistaxis, menorrhagia, easy bruising, gingival bleeding, and prolonged bleeding post-procedure.
- Initial investigations: FBC, blood film, APTT, PT/INR, fibrinogen, VWF antigen (VWF:Ag), VWF activity (VWF:GPIbM or VWF:RCo), Factor VIII:C, and VWF multimer analysis.
- VWF:Ag/VWF:GPIbM ratio and multimer analysis distinguish Type 1 from Type 2 subtypes; factor VIII level helps identify Type 2N and Type 3.
- DDAVP (desmopressin) is the first-line treatment for most Type 1 VWD and some Type 2 subtypes; perform a therapeutic trial to confirm individual response.
- VWF-containing factor concentrates (e.g., Biostate®, Wilate®) are required for Type 3, severe Type 1, Type 2B, and DDAVP non-responders or refractory patients.
- Tranexamic acid is an essential adjunct for mucocutaneous bleeding control, particularly for menorrhagia, dental procedures, and epistaxis.
- Always avoid antiplatelet agents (aspirin, NSAIDs) as they worsen bleeding risk in VWD.
- Pre-procedural haemostatic planning with a haematologist is essential; aim for VWF:RCo and FVIII:C ≥50 IU/dL for major surgery.
- Iron deficiency is common due to chronic blood loss; monitor ferritin and treat with oral or IV iron as indicated.
- Aboriginal and Torres Strait Islander peoples may face barriers to diagnosis and specialist access, particularly in remote communities; culturally safe care and point-of-care testing pathways should be prioritised.
- Genetic counselling is recommended for affected families, particularly for autosomal recessive types (2N, 3).
- Pregnancy in Type 1 VWD usually shows improvement in VWF levels in the third trimester; however, Type 2 and Type 3 patients require specialist management and postpartum haemorrhage risk remains.
Introduction & Australian Epidemiology
Von Willebrand disease (VWD) is the most common inherited bleeding disorder, caused by quantitative deficiency or qualitative dysfunction of von Willebrand factor (VWF), a multimeric glycoprotein critical for platelet adhesion to damaged endothelium and stabilisation of coagulation factor VIII (FVIII) in the circulation. The prevalence of laboratory-diagnosed VWD is approximately 1% of the general population, though clinically significant bleeding occurs in only ~0.01% (1 in 10,000).
In Australia, the Australasian Society of Thrombosis and Haemostasis (ASTH) estimates that tens of thousands of Australians have some form of VWD, though many remain undiagnosed. The Royal Children's Hospital Melbourne and the Australian Bleeding Disorders Registry (managed by the National Blood Authority) are key centres for epidemiological data collection.
VWD affects males and females equally in its inheritance, though clinical presentation often differs by sex due to the haemostatic challenge of menstruation and childbirth. Women with heavy menstrual bleeding (HMB) are a particularly important diagnostic group, and delays in diagnosis remain common in Australia.
Pathophysiology
Von Willebrand factor (VWF) is a large multimeric glycoprotein synthesised by vascular endothelial cells (stored in Weibel–Palade bodies) and megakaryocytes (stored in platelet α-granules). Its haemostatic functions include:
- Platelet adhesion: VWF mediates initial platelet tethering and adhesion to exposed subendothelial collagen at sites of vascular injury, particularly under high shear stress in arterioles and capillaries.
- Platelet aggregation: VWF binds platelet GPIbα on the glycoprotein Ib–IX–V complex, facilitating platelet plug formation.
- FVIII stabilisation: VWF serves as the carrier protein for FVIII, protecting it from premature proteolytic degradation and extending its plasma half-life from ~2 hours to ~12 hours.
Deficiency or dysfunction of VWF therefore leads to two pathological consequences: impaired primary haemostasis (manifesting as mucocutaneous bleeding) and reduced FVIII levels (which may cause a secondary coagulation defect resembling mild haemophilia A). The degree of clinical bleeding depends on the severity of VWF reduction, the presence of dysfunctional VWF multimers, and the associated FVIII level.
Types & Inheritance
VWD is classified into three major types, with further subdivision of Type 2 into four subtypes (2A, 2B, 2M, 2N). Accurate typing is essential for guiding management decisions.
| Type | Mechanism | VWF:Ag | VWF:GPIbM / RCo | FVIII:C | Multimers | Inheritance |
|---|---|---|---|---|---|---|
| Type 1 | Partial quantitative deficiency of VWF | ↓ | ↓ (proportionate) | ↓ or low-normal | Normal pattern | AD (variable penetrance) |
| Type 2A | Decreased high-molecular-weight (HMW) multimers; defective secretion or increased ADAMTS13 proteolysis | ↓ or N | ↓↓ (disproportionate) | ↓ or N | Absent HMW multimers | AD |
| Type 2B | Enhanced binding to GPIb → loss of HMW multimers and possible thrombocytopaenia | ↓ or N | ↓↓ (disproportionate) | ↓ or N | Absent HMW; enhanced platelet agglutination with ristocetin | AD |
| Type 2M | Decreased platelet-dependent function despite normal multimer distribution | N or ↓ | ↓↓ (disproportionate) | N or ↓ | Normal pattern | AD |
| Type 2N | Defective FVIII binding; mimics mild haemophilia A | N | N | ↓↓ | Normal pattern | AR |
| Type 3 | Virtual absence of VWF (homozygous or compound heterozygous null alleles) | Absent / <3 IU/dL | Absent | <10 IU/dL | Absent | AR |
Type 1 — Partial Quantitative Deficiency (~70–80% of VWD)
Type 1 is the most common form and is inherited in an autosomal dominant pattern with variable penetrance and expressivity. VWF levels may range from ~20–50 IU/dL (mild) to <10 IU/dL (severe). Blood group O individuals have ~25% lower baseline VWF levels, which can confound diagnosis. VWF:Ag and VWF:RCo are reduced proportionately, and multimer analysis shows a normal distribution.
Phenotypic expression can be influenced by ABO blood group, age, stress, exercise, inflammation, and hormonal status (e.g., pregnancy, oral contraceptives). Patients with Type 1 VWD are typically excellent responders to DDAVP.
Type 2 — Qualitative Defects (~15–20% of VWD)
Type 2 VWD is subdivided into four functional variants. In all Type 2 subtypes, there is a discrepancy between VWF antigen level and functional activity (VWF:RCo/VWF:Ag ratio <0.7 is a key diagnostic clue).
- Type 2A: The most common qualitative subtype. Characterised by loss of high-molecular-weight (HMW) multimers due to either defective intracellular processing or enhanced ADAMTS13-mediated proteolysis. Results in impaired platelet adhesion. Usually autosomal dominant.
- Type 2B: Gain-of-function mutations in the GPIbα-binding domain of VWF lead to spontaneous binding to platelets. HMW multimers are consumed, and thrombocytopaenia may occur, especially during physiological stress, pregnancy, or infection. The RIPA (ristocetin-induced platelet aggregation) assay shows increased sensitivity.
- Type 2M: Loss-of-function mutations that impair VWF binding to platelet GPIb without affecting multimer assembly. Multimer pattern is normal but function is reduced. Autosomal dominant.
- Type 2N: Mutations in the FVIII-binding site of VWF lead to failure to stabilise FVIII, resulting in low FVIII:C with normal VWF levels. This subtype mimics mild haemophilia A and is autosomal recessive. A specific FVIII binding assay or genetic testing is needed to differentiate from haemophilia A in males, and can be critical for genetic counselling.
Type 3 — Virtual Absence of VWF (<5% of VWD)
Type 3 VWD is the most severe form, inherited in an autosomal recessive pattern. Patients have virtually undetectable VWF:Ag (<3 IU/dL) and FVIII:C levels usually <10 IU/dL. Clinical features include severe mucocutaneous bleeding, haemarthroses, and soft-tissue haematomata resembling moderate-to-severe haemophilia A. Patients do not respond to DDAVP and require VWF-containing concentrates for haemostasis.
Clinical Features — Mucocutaneous Bleeding
The hallmark of VWD is mucocutaneous bleeding due to defective primary haemostasis. Symptoms vary by VWD type and severity but share common features:
Common Clinical Manifestations
| Symptom | Prevalence | Clinical Notes |
|---|---|---|
| Epistaxis | ~60–70% | Most common presenting symptom; often recurrent from childhood |
| Heavy menstrual bleeding (HMB) | ~50–90% of women | Leading cause of iron deficiency; may be first presentation in adolescence |
| Easy/prolonged bruising | ~50–60% | Spontaneous ecchymoses, particularly on limbs |
| Prolonged bleeding post-procedure | ~40–50% | Dental extraction, tonsillectomy, circumcision, surgery |
| Postpartum haemorrhage | ~20–40% | Primary and secondary PPH; VWF levels fall postpartum |
| Gingival bleeding | ~30–40% | Spontaneous or post-brushing; respond well to tranexamic acid mouthwash |
| GI bleeding | ~10–15% | May be occult; angiodysplasia more common in Type 2A |
| Haemarthroses / muscle haematomata | Type 3 only | Due to very low FVIII:C levels (<10 IU/dL) |
Investigations
Diagnosis of VWD requires a combination of clinical history, family history, and a panel of specialised laboratory tests. Testing should be performed when the patient is well and not acutely bleeding, as VWF levels are elevated by stress, inflammation, pregnancy, and oral contraceptive use.
Initial Screening Tests
Specialised VWD Diagnostic Panel
Genetic Testing
VWF gene sequencing (locus 12p13.31) is available through specialised laboratories in Australia, including through the Australian Genomics Health Alliance. It is most useful for:
- Confirming diagnosis when laboratory results are ambiguous
- Distinguishing Type 2N VWD from mild haemophilia A
- Family studies and genetic counselling for recessive types
- Predicting DDAVP response in borderline cases
DDAVP Trial
A therapeutic trial with desmopressin (DDAVP) is recommended for all newly diagnosed Type 1 patients and select Type 2 patients (not Type 2B). This confirms individual responsiveness:
- Protocol: DDAVP 0.3 μg/kg IV infusion over 20–30 minutes (or 300 μg intranasal for adults >50 kg)
- Measure: VWF:Ag, VWF:RCo/GPIbM, and FVIII:C at baseline, 1 hour, and 4 hours post-DDAVP
- Adequate response: ≥3-fold rise in VWF:RCo and FVIII:C to ≥50 IU/dL
- Monitor: Hyponatraemia risk (fluid restrict post-infusion), tachyphylaxis with repeated dosing
Risk Stratification & Severity Scoring
Severity assessment guides the intensity of haemostatic cover and the setting of management:
Procedure-Related Risk
| Procedure Risk | Examples | Target VWF:RCo / FVIII:C | Duration |
|---|---|---|---|
| Minor | Dental extraction, skin biopsy, minor endoscopy | ≥50 IU/dL | Single dose often sufficient + tranexamic acid |
| Moderate | Tonsillectomy, colonoscopy with polypectomy, arthroscopy | ≥50–70 IU/dL | 3–5 days |
| Major | Abdominal surgery, joint replacement, caesarean section | ≥80–100 IU/dL (peak), maintain ≥50 IU/dL | 7–14 days |
Management
Management of VWD involves both on-demand treatment of bleeding episodes and prophylactic cover for procedures. Three main pharmacological agents are used, often in combination.
First-Line Agents
Additional Agents & Adjuncts
Management Algorithm by VWD Type
Monitoring
- VWF and FVIII levels: Monitor VWF:Ag, VWF:RCo/GPIbM, and FVIII:C at baseline and annually in stable patients; more frequently during treatment for acute bleeding or surgery.
- Post-DDAVP levels: Measure 1 and 4 hours post-DDAVP to confirm response; repeat with each therapeutic course if using intermittently.
- Post-concentrate levels: VWF:RCo and FVIII:C at 15–30 minutes post-infusion (peak recovery) and pre-dose (trough). Adjust dosing to maintain targets.
- FVIII accumulation: With repeated VWF concentrate dosing, FVIII:C can rise above target (risk of thrombosis). Monitor daily FVIII:C during prolonged treatment courses.
- Iron studies: Ferritin, transferrin saturation, and FBC every 3–6 months if chronic blood loss; more frequently if symptomatic anaemia.
- Sodium: Serum sodium 12–24 hours post-DDAVP, especially in children, elderly, and those receiving repeated doses.
- Inhibitor screen: Consider VWF inhibitor testing in Type 3 patients who show poor response to concentrate therapy or experience anaphylaxis during infusion.
- Bleeding diary: Encourage patients to maintain a bleeding symptom diary for ongoing clinical assessment and treatment adjustment.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Laffan MA, Lester W, O'Donnell JS, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014;167(4):453–465.
- 2. Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14(2):171–232.
- 3. Connell NT, Flood VH, Brignardello-Petersen R, et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. 2021;5(1):301–325.
- 4. Australasian Society of Thrombosis and Haemostasis (ASTH). Guidelines for the management of inherited bleeding disorders in Australia and New Zealand. Intern Med J. 2020;50(Suppl 1):1–48.
- 5. National Blood Authority Australia. National Blood Authority Annual Report 2022–23. Canberra: NBA; 2023.
- 6. Favaloro EJ, Pasalic L, Curnow J. Laboratory tests used to help diagnose von Willebrand disease: an update. Pathology. 2016;48(4):303–315.
- 7. Favaloro EJ, Bonar R, Favaloro J, et al. Blood group O individuals have reduced VWF:Ag and VWF:RCo levels: implications for diagnosis of von Willebrand disease in Australia. Pathology. 2011;43(6):589–594.
- 8. Haemophilia Foundation Australia. Von Willebrand Disease: A Guide for Patients and Families. Melbourne: HFA; 2022.
- 9. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2020 summary report. Canberra: AIHW; 2020.
- 10. James AH, Kouides PA, Abdul-Kadir R, et al. Evaluation and management of acute menorrhagia in women with and without underlying bleeding disorders: consensus from an international expert panel. Eur J Obstet Gynecol Reprod Biol. 2011;158(2):124–134.
- 11. Castaman G, Goodeve A, Eikenboom J; European Group on von Willebrand Disease. Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica. 2013;98(5):667–674.
- 12. Rodeghiero F, Castaman G, Tosetto A, et al. The discriminant power of bleeding history for the diagnosis of type 1 von Willebrand disease: an international, multicenter study. J Thromb Haemost. 2005;3(12):2619–2626.
- 13. Franchini M, Mannucci PM. Von Willebrand disease–associated angiodysplasia: a few answers, still many questions. Br J Haematol. 2013;161(2):177–182.
- 14. Kadir RA, Economides DL, Sabin CA, et al. Frequency of inherited bleeding disorders in women with menorrhagia. Lancet. 1998;351(9101):485–489.