📋 Key Information Summary
- Immune thrombocytopenia (ITP) is an autoimmune disorder characterised by isolated thrombocytopenia (platelet count <100 × 10⁹/L) in the absence of other causes of thrombocytopenia — it is a diagnosis of exclusion.
- Primary mechanism: IgG autoantibodies (predominantly against GPIIb/IIIa and GPIb/IX) opsonise platelets for Fc-receptor-mediated splenic destruction; impaired megakaryopoiesis contributes in most patients.
- Incidence in Australia is approximately 3.3 per 100 000 adults per year; children 1.9–6.4 per 100 000, with a male:female ratio approaching 1:1 in adults but slight male predominance in children.
- Clinical presentation ranges from incidental asymptomatic thrombocytopenia to life-threatening mucocutaneous and intracranial haemorrhage; platelet count does not always correlate with bleeding risk.
- Treatment is indicated for platelet count <30 × 10⁹/L, significant mucocutaneous bleeding, or prior to procedures — NOT for the count alone when asymptomatic above threshold.
- First-line therapy: corticosteroids — prednis(ol)one 1 mg/kg/day (max 80 mg) PO for 1–2 weeks then taper over 2–4 weeks, OR pulsed dexamethasone 40 mg PO daily × 4 days (1–3 cycles).
- For emergency bleeding or urgent pre-procedural platelet rise: IV immunoglobulin (IVIg) 0.4–1 g/kg over 1–5 days ± platelet transfusion with concurrent IVIg or tranexamic acid.
- Second-line therapy for chronic/refractory ITP: thrombopoietin receptor agonists (TPO-RAs) — romiplostim (SC weekly) or eltrombopag (PO daily) — are PBS Authority Required and achieve response in 60–90% of patients.
- Rituximab (anti-CD20 monoclonal antibody) offers 60% initial response rate but ~50% relapse by 1–2 years; used off-PBS in Australia for steroid-refractory ITP.
- Splenectomy remains curative in ~60–70% but is deferred until ≥12 months of medical therapy and avoided in children <5 years due to overwhelming infection risk.
- Children: 75–80% of paediatric ITP is self-limiting within 6–12 months; watchful waiting appropriate if platelet count >20 × 10⁹/L and no significant bleeding.
- ATSI Australians have higher rates of ITP complications and reduced access to haematology specialist care; engage MBS telehealth item 91822 and ensure culturally safe management.
Introduction & Australian Epidemiology
Immune thrombocytopenia (ITP) is an acquired autoimmune disorder defined by a platelet count below 100 × 10⁹/L in the absence of other identifiable causes of thrombocytopenia. It may present as an isolated haematological finding or as part of a systemic condition. The International Working Group (IWG) standardised terminology in 2009, replacing the older term "idiopathic thrombocytopenic purpura" with "immune thrombocytopenia" to reflect the autoimmune pathogenesis.
In Australia, population-based studies estimate adult ITP incidence at 3.3 per 100 000 per year, with a prevalence of approximately 9.5 per 100 000. Paediatric ITP peaks between ages 2–5 years with an annual incidence of 1.9–6.4 per 100 000. Unlike many autoimmune conditions, adult ITP shows an approximately equal sex distribution, though some series report a slight female preponderance.
ITP is classified as primary (isolated, no associated condition) in approximately 80% of adults and 70% of children, and secondary when associated with conditions such as systemic lupus erythematosus (SLE), antiphospholipid syndrome, chronic lymphocytic leukaemia, hepatitis C, HIV, or Helicobacter pylori infection. Secondary causes must be actively excluded during initial workup.
The disease course is defined as newly diagnosed (<3 months), persistent (3–12 months), or chronic (>12 months). In adults, ~60% become chronic; in children, 75–80% undergo spontaneous remission within 6–12 months, making the prognosis markedly more favourable in the paediatric population.
Mortality in ITP is predominantly driven by bleeding (intracranial haemorrhage rate ~0.1–0.5% overall, but ~5% in refractory severe ITP) and infection (especially post-splenectomy overwhelming infection). A landmark Danish registry study demonstrated increased all-cause mortality in ITP patients compared with the general population (standardised mortality ratio 1.5–2.2), with age, refractory disease, and treatment-related complications as major drivers.
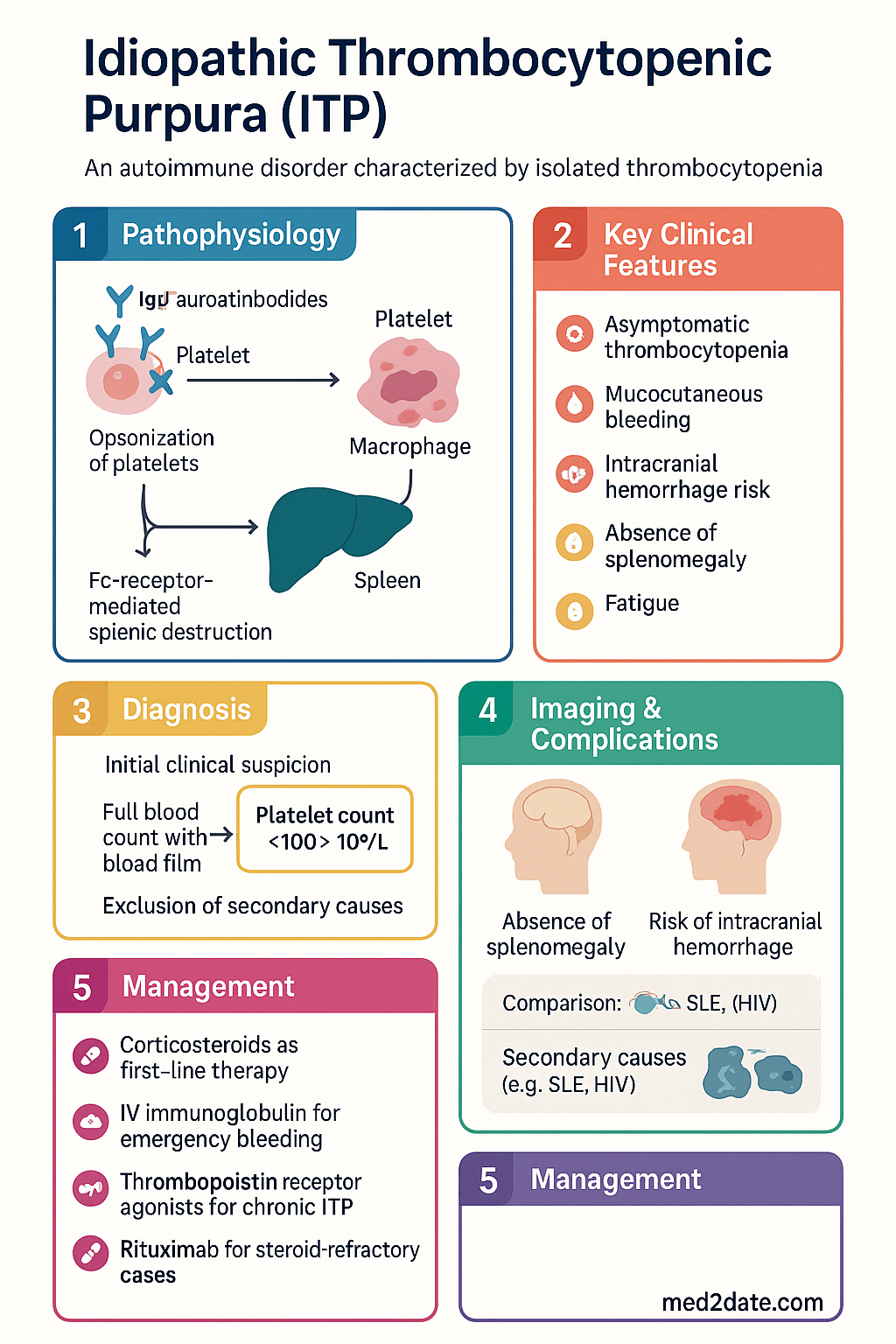
Pathogenesis: Anti-Platelet IgG Antibodies
The pathophysiology of ITP involves a complex interplay of humoral immune dysregulation, cell-mediated cytotoxicity, and impaired thrombopoiesis. The traditional view of ITP as a pure antibody-mediated platelet destruction disorder has evolved into a multi-hit model.
Antibody-Mediated Platelet Destruction
In 50–75% of patients, IgG autoantibodies target platelet surface glycoproteins, most commonly:
- GPIIb/IIIa (integrin αIIbβ3) — the most frequent target (~70% of antibody-positive cases); also expressed on megakaryocytes, so antibodies may directly inhibit platelet production.
- GPIb/IX/V (von Willebrand factor receptor) — second most common (~20–40%); GPIb-directed antibodies may cause Fc-independent platelet clearance via desialylation and hepatic Ashwell–Morell receptor uptake.
- GPIa/IIa (collagen receptor) and GPIV (CD36) — less frequent; present in <10% of cases individually.
Opsonised platelets are cleared predominantly in the spleen by Fcγ receptor-bearing macrophages in the red pulp. The spleen also serves as the major site of autoantibody production by long-lived plasma cells and memory B cells resident in the marginal zone.
Cell-Mediated Immune Mechanisms
Patients seronegative for anti-platelet antibodies (25–50% of ITP patients) have demonstrable T-cell-mediated mechanisms:
- Th1/Th17 skewing with reduced Treg function and expanded cytotoxic CD8⁺ T cells capable of direct platelet and megakaryocyte lysis.
- Reduced production of IL-10 and TGF-β by regulatory T cells, permitting perpetuation of autoimmune responses.
- Increased platelet apoptosis driven by caspase activation, independent of splenic sequestration.
Impaired Thrombopoiesis
Anti-platelet antibodies cross-react with megakaryocyte surface glycoproteins (particularly GPIIb/IIIa and GPIb/IX), inhibiting platelet budding in the bone marrow. Additionally, CD8⁺ T-cell-mediated megakaryocyte apoptosis reduces platelet production. Endogenous thrombopoietin (TPO) levels are only modestly elevated in ITP (unlike aplastic anaemia) because TPO is constitutively produced and catabolised by binding to platelet mass — a smaller platelet mass reduces TPO clearance, but the rise is insufficient to compensate for ongoing destruction.
Fcgamma Receptor Polymorphisms
FcγRIIa (CD32) and FcγRIIIa (CD16) polymorphisms influence macrophage phagocytic capacity and have been associated with ITP susceptibility and response to IVIg therapy. The H131 FcγRIIa variant shows higher affinity for IgG1 and IgG2 immune complexes and may predict more aggressive disease.
Clinical Features & Severity
The clinical presentation of ITP varies from an asymptomatic incidental finding to life-threatening haemorrhage. Bleeding severity does not always correlate with platelet count — some patients with platelet counts of 5 × 10⁹/L may have minimal bleeding, while others with counts of 30–40 × 10⁹/L may exhibit significant purpura.
Presentation by Platelet Count
Bleeding Assessment — ITP Bleeding Score (IBLS)
The ITP Bleeding Score assesses bleeding in 11 anatomical sites (skin, oral, nasal, GI, urinary, gynaecological, CNS, etc.) on a 0–4 scale per site. A total score ≥8 correlates with increased haemorrhagic risk and may prompt escalation of therapy regardless of platelet count.
Associated Findings
- Splenomegaly is typically absent in primary ITP — its presence should prompt investigation for secondary causes (lymphoproliferative disorders, portal hypertension).
- Hepatomegaly or lymphadenopathy suggest a secondary or alternative diagnosis.
- Anaemia may be present from chronic blood loss (iron deficiency) or autoimmune haemolytic anaemia (Evans syndrome — Coombs-positive in ~10–20% of ITP patients).
- Fatigue is increasingly recognised as a significant quality-of-life impairment in ITP, often disproportionate to thrombocytopenia severity and may persist even after platelet count normalisation.
Emergency Presentations
Investigations — Diagnosis of Exclusion
There is no single definitive diagnostic test for ITP. The diagnosis requires demonstration of isolated thrombocytopenia (platelet count <100 × 10⁹/L) on at least two occasions separated by >1 week, with exclusion of all other causes of thrombocytopenia. The IWG 2009 criteria form the basis of Australian diagnostic practice.
Essential Baseline Investigations
Specialised / Referral Investigations
Management: Steroids, IVIG, TPO-RA & Rituximab
Treatment decisions in ITP should be individualised based on bleeding severity, comorbidities, lifestyle factors (occupation, sport participation, planned procedures), patient preferences, and platelet count. The primary therapeutic goal is to achieve a safe platelet count (typically >30 × 10⁹/L and absence of significant bleeding), not necessarily a normal platelet count.
When to Treat
- Platelet count <30 × 10⁹/L in adults — treat regardless of symptoms
- Significant mucocutaneous bleeding at any platelet count
- Platelet count <50 × 10⁹/L with risk factors: anticoagulant therapy, planned surgery, uncontrolled hypertension, active peptic ulcer disease
- Platelet count 30–50 × 10⁹/L without bleeding — may observe with close monitoring
- In children: treatment if platelet count <20 × 10⁹/L with bleeding symptoms, or <10 × 10⁹/L regardless of symptoms
First-Line Therapy
Corticosteroids
IV Immunoglobulin (IVIg)
Second-Line Therapy
Second-line agents are indicated when patients have persistent or chronic ITP (≥3–6 months duration) and meet any of: platelet count persistently <30 × 10⁹/L despite first-line therapy, recurrent significant bleeding, intolerable steroid side effects, or need for ongoing therapy to maintain safe counts.
Thrombopoietin Receptor Agonists (TPO-RAs)
Rituximab
Third-Line / Refractory ITP
Approximately 10–20% of ITP patients are refractory to first- and second-line therapies. Options include:
- Splenectomy: Laparoscopic splenectomy offers sustained complete response in 60–70%. Pre-operative vaccination essential: pneumococcal (Pneumovax 23® + Prevenar 13® at least 2 weeks prior, ideally 6 weeks), meningococcal (Nimenrix® — MenACWY), Haemophilus influenzae type b. Lifelong post-splenectomy antibiotic prophylaxis with phenoxymethylpenicillin 250 mg PO BD (or 500 mg daily) recommended by RACGP guidelines.
- Combination therapy: TPO-RA + rituximab may achieve higher and more durable responses than either agent alone (SYNERGY study ongoing).
- Mycophenolate mofetil: 1 g PO BD — emerging evidence as steroid-sparing agent; response rate ~40–60% in refractory ITP.
- Azathioprine: 2 mg/kg/day PO — slow onset (3–6 months); response rate ~40–50%. PBS General Benefit.
- Cyclosporin: 2–3 mg/kg/day PO — limited evidence; used in refractory cases. Monitor renal function and blood pressure.
- Danazol: 200 mg PO TDS — androgen; response in ~40–60%; hepatotoxicity monitoring required. Limited availability in Australia.
Emergency Management of Life-Threatening Bleeding
Monitoring
| Agent | Monitoring | Frequency |
|---|---|---|
| Corticosteroids | Blood glucose, blood pressure, weight, bone density (if >3 months use) | Weekly during treatment; DEXA if prolonged |
| IVIg | Renal function, haemolysis markers (DAT, LDH, haptoglobin), fluid balance | Before each infusion; 24–48 h post if symptoms |
| Romiplostim | FBC with platelet count, peripheral blood film (reticulin), thromboembolic symptoms | Weekly during titration; monthly when stable; blood film every 6–12 months |
| Eltrombopag | LFTs, FBC, ophthalmological exam (cataracts), iron studies | LFTs fortnightly × 3 months then monthly; eye exam every 6 months |
| Rituximab | Immunoglobulin levels (IgG, IgA, IgM), HBV DNA, CD19 B-cell count | IgG at 3-monthly; HBV DNA if carrier; CD19 at 3–6 months post-treatment |
| Post-splenectomy | FBC (for target Howell–Jolly bodies), infection surveillance, vaccination status | FBC annually; lifelong fever monitoring (<38°C = immediate medical review) |
Special Populations
Pregnancy
Paediatrics
Elderly (≥65 years)
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal and Torres Strait Islander Health Considerations
Quick Reference: ITP Treatment Algorithm
📚 References
- 1. Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780–3817. doi:10.1182/bloodadvances.2019000812
- 2. Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829–3866. doi:10.1182/bloodadvances.2019000966
- 3. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–2393. doi:10.1182/blood-2008-07-162503
- 4. Mithoowani S, Gregory-Miller K, Goy J, et al. High-dose dexamethasone compared with standard-dose prednisone for initial treatment of primary immune thrombocytopenia: a systematic review and meta-analysis. Lancet Haematol. 2016;3(10):e489–e496. doi:10.1016/S2352-3026(16)30103-3
- 5. Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet. 2011;377(9763):393–402. doi:10.1016/S0140-6736(10)60959-2
- 6. Tarantino MD, Bussel JB, Blanchette VS, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388(10039):45–54. doi:10.1016/S0140-6736(16)00279-8
- 7. Arnold DM, Heddle NM, Cook RJ, et al. Reduced-dose rituximab in adults with immune thrombocytopenia: a randomised controlled trial (GODEFRIDUS). Blood. 2023;142(Supplement 1):67. doi:10.1182/blood-2023-181748
- 8. Frederiksen H, Maegbaek ML, Norgaard M. Twenty-year mortality of adult patients with primary immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol. 2014;166(2):260–267. doi:10.1111/bjh.12869
- 9. Grace RF, Despotovic JM, Bennett CM, et al. Eltrombopag in children with immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet. 2018;391(10115):30–38. doi:10.1016/S0140-6736(17)32859-4
- 10. Australian Government Department of Health. National Blood Authority. National Guidelines for the Use of Immunoglobulin in Australia. 2023. Available at: www.blood.gov.au
- 11. RANZCOG (Royal Australian and New Zealand College of Obstetricians and Gynaecologists). Management of Thrombocytopenia in Pregnancy. College Statement C-Obs 67. 2022.
- 12. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023. Canberra: AIHW; 2023.
- 13. Australasian Society for Clinical Immunology and Allergy (ASCIA), RACGP. Australian Immunisation Handbook: Post-Splenectomy Vaccination and Prophylaxis. Australian Government Department of Health; 2023.
- 14. Ghanima W, Godeau B, Cines DB, Bussel JB. How I treat immune thrombocytopenia: the choice between splenectomy or a medical therapy as a second-line treatment. Blood. 2012;120(5):960–969. doi:10.1182/blood-2011-12-309153