📋 Key Information Summary
- TTP is a medical emergency — untreated mortality exceeds 90%; early plasma exchange (PEX) reduces mortality to 10–20%.
- Thrombotic microangiopathy (TMA) driven by severe ADAMTS13 deficiency (activity <10%) leading to ultra-large von Willebrand factor (vWF) multimer accumulation and microvascular thrombosis.
- The classic pentad — thrombocytopenia, microangiopathic haemolytic anaemia (MAHA), renal impairment, neurological features, and fever — is present in its entirety in <10% of patients; do not wait for all five features before initiating PEX.
- A presumptive diagnosis of TTP should be made whenever MAHA and thrombocytopenia are present without another obvious cause (e.g., DIC, HUS, HELLP).
- Therapeutic plasma exchange (PEX) is first-line treatment — removes autoantibodies and ultra-large vWF multimers while replacing ADAMTS13 enzyme.
- Caplacizumab (anti-vWF nanobody) reduces time to platelet normalisation and TTP-related death/organ failure; PBS Authority Required listing in Australia.
- Rituximab (anti-CD20) is second-line for refractory/relapsing TTP and increasingly used as upfront adjunctive therapy in high-risk patients.
- ADAMTS13 activity <10% with positive anti-ADAMTS13 IgG confirms acquired TTP; a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13 (ADAMTS13) assay turnaround is typically 5–7 days in Australia.
- The PLASMIC score or French score can risk-stratify patients for probable severe ADAMTS13 deficiency while awaiting results.
- Congenital TTP (Upshaw–Schulman syndrome) requires fresh frozen plasma infusions rather than PEX and is managed with a haematologist.
- Relapse occurs in 30–50% of acquired TTP; maintenance rituximab and serial ADAMTS13 monitoring guide pre-emptive therapy.
- ATSI Australians have higher TMA incidence and face barriers to timely PEX access — early retrieval coordination via RFDS is essential.
Introduction & Australian Epidemiology
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening thrombotic microangiopathy (TMA) characterised by severe deficiency of the von Willebrand factor (vWF)-cleaving protease ADAMTS13. Inadequate cleavage of ultra-large vWF multimers results in widespread platelet-rich microthrombi, consumptive thrombocytopenia, and mechanical haemolysis of red blood cells.
TTP accounts for approximately 15–20% of all TMA presentations in Australian tertiary centres. The incidence of acquired TTP in Australia is estimated at 3–11 cases per million per year, with a female predominance (approximately 3:1) and peak incidence in the 30–50-year age group. Aboriginal and Torres Strait Islander peoples may have a higher burden of TMA-related illness, partly driven by higher rates of autoimmune comorbidities and delayed access to specialist care in remote settings.
Acquired TTP is autoimmune, driven by inhibitory IgG autoantibodies against ADAMTS13. Congenital TTP (Upshaw–Schulman syndrome, OMIM #274150) is rare, autosomal recessive, and accounts for <5% of cases. Triggers for acquired TTP include pregnancy, autoimmune disease (particularly SLE), infections, and certain medications (e.g., ticlopidine, clopidogrel — rare).
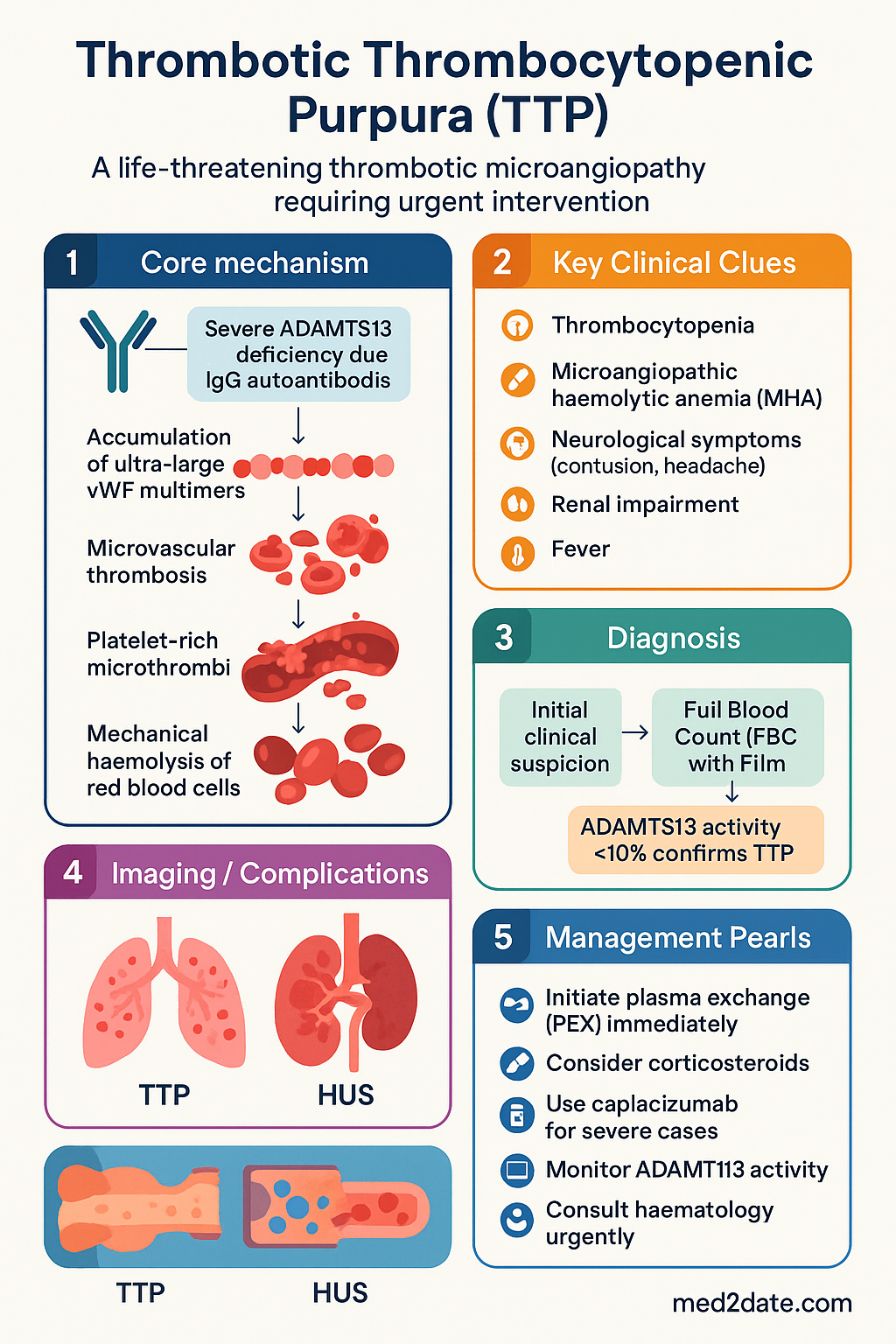
Pathogenesis — ADAMTS13 Deficiency
ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13) is a zinc metalloprotease synthesised predominantly by hepatic stellate cells. Its physiological role is to cleave ultra-large (UL) vWF multimers secreted by endothelial cells into smaller, less thrombogenic forms.
Mechanism of Acquired (Autoimmune) TTP
- IgG autoantibodies (predominantly IgG1 and IgG4) bind ADAMTS13, causing functional inhibition and/or accelerated clearance via the reticuloendothelial system.
- ADAMTS13 activity falls to <10% of normal, resulting in accumulation of UL-vWF multimers in the microvasculature.
- Under high shear stress (particularly arteriolar), UL-vWF unfolds and recruits platelets via glycoprotein Ibα (GPIbα), forming platelet-rich microthrombi.
- These thrombi mechanically fragment red blood cells (schistocytes), producing MAHA.
- Consumptive thrombocytopenia results from platelet incorporation into microthrombi.
- End-organ ischaemia produces the clinical features: brain (neurological symptoms), kidney (renal impairment), and systemic (fever).
Congenital TTP (Upshaw–Schulman Syndrome)
- Biallelic mutations in the ADAMTS13 gene (chromosome 9q34) lead to absent or severely reduced ADAMTS13 protein.
- Typically presents in neonatal period or early childhood with jaundice, thrombocytopenia, and haemolysis — often triggered by intercurrent infection.
- No autoantibodies detected; genetic testing confirms the diagnosis.
- Managed with prophylactic plasma infusions (10–15 mL/kg every 2–3 weeks) to supply exogenous ADAMTS13.
Distinguishing TTP from Other TMAs
| Feature | TTP | HUS (typical/complement) | DIC |
|---|---|---|---|
| ADAMTS13 activity | <10% | Normal or mildly reduced | Normal |
| Platelets | Very low (often <30) | Low–moderate | Low |
| Renal failure | Mild–moderate | Severe (hallmark) | Variable |
| Schistocytes | Present | Present | May be present |
| Coagulation screen | Normal (PT/APTT) | Normal | Prolonged PT/APTT |
| First-line treatment | Plasma exchange | Supportive ± eculizumab | Treat underlying cause |
Clinical Pentad & Presentation
The traditional pentad of TTP comprises five clinical features. However, the complete pentad is present in only 5–10% of patients at presentation. Most patients present with the triad of thrombocytopenia, MAHA, and non-specific neurological symptoms. A high index of suspicion is required.
Predisposing Factors & Triggers
- Female sex (3:1 female predominance, likely oestrogen-related immune modulation)
- African ancestry (higher autoantibody prevalence; relevant in multicultural Australian populations)
- Systemic lupus erythematosus and other autoimmune conditions
- Pregnancy (especially 2nd and 3rd trimester — must differentiate from pre-eclampsia/HELLP)
- HIV infection
- Drugs: ticlopidine, clopidogrel (rare), quinine, ciclosporin
- Bone marrow transplantation
Investigations
Laboratory confirmation of TTP requires demonstration of thrombocytopenia, MAHA (with schistocytes), and critically, severe ADAMTS13 deficiency. While awaiting ADAMTS13 results (typically 5–7 working days in Australian reference laboratories), treatment must not be delayed.
Essential & Urgent Investigations
Risk-Stratification Scores
While awaiting ADAMTS13 results, clinical scoring systems help estimate the probability of severe ADAMTS13 deficiency and guide early treatment decisions.
PLASMIC Score
| Parameter | Points |
|---|---|
| Platelet count <30 × 10⁹/L | 1 |
| Haemolysis (reticulocytes >2.5%, haptoglobin undetectable, indirect bilirubin >2 mg/dL) | 1 |
| No active cancer | 1 |
| No stem-cell or solid organ transplant | 1 |
| MCV <90 fL | 1 |
| INR <1.5 | 1 |
| Creatinine <2.0 mg/dL (176 µmol/L) | 1 |
Interpretation: Score ≥5 → high probability of severe ADAMTS13 deficiency (PPV ~84%). Score 0–4 → low probability. A high PLASMIC score in the setting of MAHA and thrombocytopenia should strongly support immediate PEX initiation.
Risk Stratification & Severity Scoring
Management — Plasma Exchange, Rituximab & Caplacizumab
Immediate Management (First 24 Hours)
Therapeutic Plasma Exchange (PEX)
PEX remains the cornerstone of TTP treatment and should be initiated within 4–8 hours of clinical suspicion. PEX performs three critical functions simultaneously:
- Removes pathogenic anti-ADAMTS13 autoantibodies
- Removes ultra-large vWF multimers
- Replaces functional ADAMTS13 enzyme via donor FFP
| Parameter | Detail |
|---|---|
| Volume exchanged | 1.0–1.5 × calculated plasma volume daily |
| Replacement fluid | Fresh frozen plasma (FFP) or cryoprecipitate-poor plasma (cryo-poor FFP — preferentially used in some centres) |
| Frequency | Daily until platelet count >150 × 10⁹/L for ≥2 consecutive days and LDH normalised |
| Taper | After initial response: reduce to alternate days, then twice weekly, then cease over 1–2 weeks |
| Refractory definition | No platelet count improvement after 5–7 days of daily PEX — escalate: twice-daily PEX + rituximab |
| Venous access | Large-bore central venous catheter (e.g., Mahurkar dialysis catheter) inserted by experienced operator |
Caplacizumab (Cablivi®)
Caplacizumab is a humanised anti-vWF A1-domain nanobody that blocks the interaction between UL-vWF multimers and platelet glycoprotein Ibα (GPIbα), preventing microthrombus formation independently of ADAMTS13 reconstitution.
Rituximab
Rituximab depletes CD20+ B lymphocytes, reducing anti-ADAMTS13 autoantibody production. Used for refractory, relapsing, or high-risk acquired TTP.
Corticosteroids
Refractory TTP — Second-Line & Salvage Options
| Agent | Mechanism | Dose | Notes |
|---|---|---|---|
| Rituximab (if not yet used) | Anti-CD20 B-cell depletion | 375 mg/m² IV weekly × 4 | Standard second-line; response in 80–90% refractory cases |
| Bortezomib | Proteasome inhibitor — targets plasma cells | 1.3 mg/m² SC/IV Days 1, 4, 8, 11 (21-day cycle) | Case-series evidence. Effective when rituximab fails. Specialist-only. PBS authority may be needed. |
| Cyclophosphamide | Alkylating agent — broad immunosuppression | 500–1000 mg/m² IV single dose or monthly | Rarely used; for multiply-refractory TTP. Haematology/oncology supervision required. |
| Vincristine | Microtubule inhibitor | 2 mg IV weekly × 4 | Historical salvage option; limited evidence. Use only when other options exhausted. |
| Splenectomy | Removes antibody-producing and vWF-clearing organ | N/A — surgical | Last resort for multiply-refractory TTP. Limited to case series. |
Monitoring During Treatment
- Platelet count: Daily until >150 × 10⁹/L for ≥2 days, then taper PEX frequency.
- LDH: Daily during active treatment — should fall with platelet recovery.
- FBC, haemolysis markers: Daily during PEX, then 2–3 times weekly during taper.
- ADAMTS13 activity: Repeat at Day 7–14, then monthly for 6–12 months to guide rituximab maintenance and detect impending relapse.
- Troponin: Baseline and if chest pain/dyspnoea — TTP-related myocardial microthrombi cause elevated troponin without epicardial coronary disease.
- Neurological status: Serial neurological examinations; fluctuations are common and may indicate active disease.
- Bleeding: Particularly on caplacizumab — monitor for epistaxis, gingival bleeding, GI bleeding. Hold caplacizumab for platelet count >50 × 10⁹/L with active bleeding.
- Immunoglobulin levels: After rituximab — monitor IgG monthly; IVIg replacement if IgG <4 g/L with recurrent infections.
When to Stop PEX
- Platelet count >150 × 10⁹/L for ≥2 consecutive days AND LDH normalised — begin PEX taper.
- Taper: alternate day × 3 sessions, then twice weekly × 2 sessions, then cease.
- Caplacizumab continues for 30 days after last daily PEX.
- If platelets fall during taper → increase PEX frequency back to daily.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Thrombotic microangiopathies, including TTP, present particular challenges for Aboriginal and Torres Strait Islander communities. The following considerations are essential for equitable management.
📚 References
- 1. Scully M, Cataland SR, Peyvandi F, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335–346.
- 2. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836–2846.
- 3. Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2496–2502.
- 4. Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2486–2495.
- 5. Bendapudi PK, Hurwitz S, Fry A, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies. Lancet Haematol. 2017;4(4):e157–e164.
- 6. Coppo P, Bubenheim M, Azoulay E, et al. A regimen with caplacizumab, immunosuppression, and plasma exchange prevents unfavorable outcomes in immune-mediated TTP. Blood. 2021;137(6):733–742.
- 7. Sayani FA, Abrams CS. How I treat refractory thrombotic thrombocytopenic purpura. Blood. 2015;125(25):3860–3867.
- 8. Dutt T, Shaw RJ, Stubbs M, et al. Real-world experience with caplacizumab in the management of acute TTP. Br J Haematol. 2021;194(5):e118–e121.
- 9. Patriquin CJ, Thomas MR, Dutt T, et al. Bortezomib in the treatment of refractory thrombotic thrombocytopenic purpura. Br J Haematol. 2016;173(5):779–785.
- 10. National Blood Authority Australia. Australian National Blood Authority Patient Blood Management Guidelines: Module 5 — Patients Requiring Massive Transfusion. Canberra: NBA; 2018.
- 11. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2020 Summary Report. Canberra: AIHW; 2020.
- 12. Kremer Hovinga JA, Coppo P, Lämmle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers. 2017;3:17020.
- 13. Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. 2017;1(10):590–600.
- 14. Riva S, Mancini I, Maino A, et al. Long-term follow-up of idiopathic TTP: a cohort study. Blood Adv. 2020;4(7):1472–1480.
- 15. Royal Flying Doctor Service of Australia. Retrieval and Emergency Coordination Guidelines. RFDS; 2023.