📋 Key Information Summary
- CLL is the most common adult leukaemia in Australia, with median age at diagnosis 70–72 years; characterised by clonal proliferation of mature B lymphocytes.
- Staging determines prognosis and treatment urgency: Rai (0–IV) and Binet (A–C) systems remain the backbone of clinical staging and are widely used in Australian haematology practice.
- Most patients are asymptomatic at diagnosis — incidental lymphocytosis on FBC is the commonest presentation; up to 70% of new diagnoses are early-stage (Rai 0/I, Binet A).
- Watch-and-wait (active surveillance) is standard of care for early-stage, asymptomatic CLL; no evidence that early treatment of asymptomatic disease improves survival.
- Immunophenotyping is essential for diagnosis: CD5+/CD23+/CD20(dim)/sIg(dim) co-expression on flow cytometry; clonal B-cell population ≥5 × 10⁹/L in peripheral blood.
- FISH and molecular testing guide prognosis: del(17p)/TP53 mutation confers poor prognosis and resistance to chemoimmunotherapy; IGHV mutational status differentiates indolent (mutated) from aggressive (unmutated) disease.
- Treatment is indicated for symptomatic or advanced-stage disease — criteria include progressive marrow failure, massive/symptomatic splenomegaly or lymphadenopathy, lymphocyte doubling time <6 months, or progressive cytopenias.
- FCR (fludarabine, cyclophosphamide, rituximab) remains first-line for fit, younger patients with mutated IGHV and no del(17p)/TP53 aberration.
- Targeted agents have transformed CLL management: BTK inhibitors (ibrutinib, zanubrutinib) and BCL-2 inhibitors (venetoclax + obinutuzumab) are preferred for elderly, unfit, or TP53-aberrant patients — PBS-listed in Australia.
- Venetoclax + obinutuzumab offers a fixed-duration, chemotherapy-free option (12 months) with deep remissions and is PBS Authority Required for first-line CLL.
- Tumour lysis syndrome (TLS) prophylaxis is mandatory with venetoclax initiation — ramp-up dosing, hydration, and urate-lowering therapy (rasburicase or allopurinol) with electrolyte monitoring.
- Infections are the leading cause of morbidity and mortality — hypogammaglobulinaemia, neutropenia, and treatment-related immunosuppression mandate IVIg replacement and antimicrobial prophylaxis.
- CLL transformation to Richter syndrome (diffuse large B-cell lymphoma, 2–10% lifetime risk) should be suspected with rapid clinical deterioration, enlarging nodes, or rising LDH.
- Aboriginal and Torres Strait Islander patients may present later, have reduced access to specialist haematology services in remote areas, and require culturally safe care pathways; referral to urban centres for complex therapy may be necessary.
Introduction & Australian Epidemiology
Chronic lymphocytic leukaemia (CLL) is the most common adult leukaemia in the Western world, including Australia. It is characterised by the clonal accumulation of morphologically mature but immunologically incompetent B lymphocytes in the blood, bone marrow, lymph nodes, spleen, and liver. CLL has a highly indolent natural history, with many patients living for decades without requiring therapy, while others follow a more aggressive course driven by adverse cytogenetic and molecular features.
In Australia, CLL accounts for approximately 25–30% of all leukaemias, with an estimated age-standardised incidence rate of 5–7 per 100,000 per year. The median age at diagnosis is 70–72 years, and the disease has a male predominance (male-to-female ratio approximately 1.5–2:1). Incidence increases markedly with age, with the highest rates in patients aged >75 years. According to the Australian Institute of Health and Welfare (AIHW), CLL remains among the top ten most commonly diagnosed cancers in Australians over the age of 65.
Australia's geographic vastness creates unique challenges in CLL management. Patients in regional and remote areas may have limited access to haematology specialists, flow cytometry services, and FISH/molecular testing. The introduction of oral targeted therapies (ibrutinib, venetoclax, zanubrutinib) has improved access to treatment outside tertiary centres, although PBS authority requirements, specialised monitoring (e.g., venetoclax TLS protocol), and infectious disease complications still necessitate coordinated multidisciplinary care.
The past two decades have witnessed a paradigm shift in CLL management — from chemoimmunotherapy-based approaches to targeted, chemotherapy-free regimens with superior survival outcomes, particularly in patients with high-risk genomic features. Australian treatment guidelines have evolved in parallel, and this article provides a comprehensive clinical framework for the diagnosis, risk stratification, and management of CLL in the Australian healthcare setting.
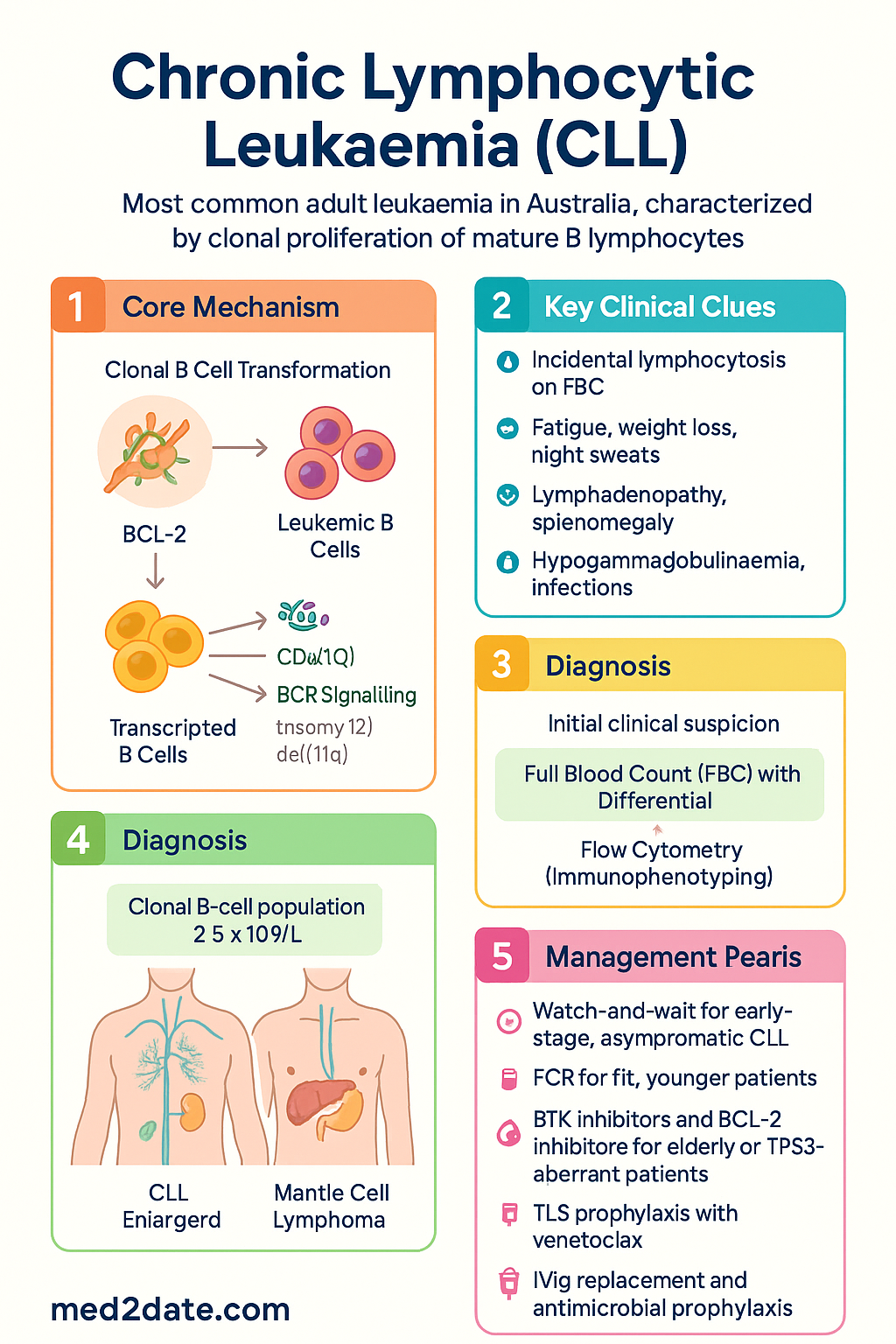
Pathogenesis & Staging (Rai/Binet)
Pathogenesis
CLL arises from a single clonal B cell that has undergone malignant transformation, typically in the germinal centre or post-germinal centre environment. The leukaemic cells share features of both naïve and memory B cells. Key pathogenic mechanisms include:
- Anti-apoptotic signalling: Overexpression of BCL-2 family proteins (BCL-2, BCL-XL, MCL-1) promotes prolonged cell survival and resistance to apoptosis.
- Microenvironment dependence: CLL cells rely on survival signals from the tumour microenvironment — nurse-like cells, T cells, and stromal cells in lymph nodes and bone marrow — via CXCR4/CXCL12, CD40/CD40L, and B-cell receptor (BCR) signalling pathways.
- BCR signalling: Constitutive or antigen-driven activation of the BCR pathway (via BTK, SYK, PI3K) is central to CLL cell proliferation and survival; this pathway is the primary target of ibrutinib and zanubrutinib.
- Cytogenetic abnormalities: Recurrent chromosomal aberrations drive clinical heterogeneity — del(13q) (most common, ~55%, favourable), trisomy 12 (~15%, intermediate), del(11q)/ATM (~18%, adverse), del(17p)/TP53 (~7%, very adverse).
- TP53 dysfunction: TP53 mutation (with or without del(17p)) confers resistance to DNA-damaging chemotherapy and is found in 5–8% of treatment-naïve CLL and 30–50% of relapsed/refractory disease.
- IGHV mutational status: Somatic hypermutation of the immunoglobulin heavy-chain variable region gene (IGHV) divides CLL into two prognostic groups — mutated IGHV (indolent, better response to chemoimmunotherapy) and unmutated IGHV (aggressive, poorer response to fludarabine-based regimens).
Rai Staging System
The Rai system, originally developed in the United States, classifies CLL into five stages based on lymphocytosis, lymphadenopathy, hepatosplenomegaly, anaemia, and thrombocytopenia. It is widely used in Australian practice:
| Rai Stage | Findings | Risk Category | Median Survival |
|---|---|---|---|
| 0 | Lymphocytosis only (blood + marrow) | Low | >10 years |
| I | Lymphocytosis + lymphadenopathy | Intermediate | >8 years |
| II | Lymphocytosis + hepatomegaly and/or splenomegaly ± lymphadenopathy | Intermediate | 6–7 years |
| III | Lymphocytosis + anaemia (Hb <110 g/L) ± organomegaly ± lymphadenopathy | High | 2–3 years (without treatment) |
| IV | Lymphocytosis + thrombocytopenia (platelets <100 × 10⁹/L) ± anaemia ± organomegaly | High | 2–3 years (without treatment) |
Binet Staging System
The Binet system, developed in Europe and also widely adopted in Australia, classifies CLL based on the number of involved lymphoid areas (cervical, axillary, inguinal lymph nodes — unilateral or bilateral — spleen, liver) and the presence of cytopenias:
| Binet Stage | Findings | Approximate Rai Equivalent |
|---|---|---|
| A | Hb ≥100 g/L, platelets ≥100 × 10⁹/L, <3 involved areas | Rai 0 |
| B | Hb ≥100 g/L, platelets ≥100 × 10⁹/L, ≥3 involved areas | Rai I–II |
| C | Hb <100 g/L and/or platelets <100 × 10⁹/L | Rai III–IV |
While clinical staging remains essential, it does not capture the biological heterogeneity of CLL. Integrating molecular prognostic factors (see Investigations) into treatment decisions is now standard of care.
Clinical Features & Complications
Presentation
The majority of CLL patients (up to 70%) are diagnosed incidentally following a routine full blood count (FBC) showing persistent, unexplained lymphocytosis. When symptomatic, the most common presentations include:
- Constitutional symptoms: Fatigue (most common), unintentional weight loss >10% over 6 months, drenching night sweats, fevers >38°C without evidence of infection (B symptoms).
- Lymphadenopathy: Painless, symmetrical, non-tender enlargement of cervical, axillary, and inguinal lymph nodes — may become massive and compress adjacent structures.
- Splenomegaly: Left upper quadrant discomfort, early satiety, or incidental finding on examination.
- Hepatomegaly: Less common than splenomegaly; generally milder.
- Signs of cytopenias: Pallor, dyspnoea, and fatigue (anaemia); petechiae, easy bruising, or mucosal bleeding (thrombocytopenia); recurrent or severe infections (neutropenia).
Complications
- Immunodeficiency and infections: The single greatest cause of morbidity and mortality in CLL. Hypogammaglobulinaemia occurs in up to 60% of patients and worsens with disease duration and treatment. Patients are susceptible to bacterial (pneumococcus, Haemophilus influenzae, Staphylococcus aureus), viral (herpes zoster reactivation, influenza, COVID-19), and fungal infections.
- Autoimmune complications: Occur in 5–10% of CLL patients. Autoimmune haemolytic anaemia (AIHA) is the most common, followed by immune thrombocytopenia (ITP) and pure red cell aplasia (PRCA). Paradoxically, autoimmune cytopenias may occur in early-stage disease and are treated with corticosteroids ± rituximab, not necessarily by initiating CLL-directed therapy.
- Richter transformation: Histological progression to aggressive lymphoma (most commonly diffuse large B-cell lymphoma, DLBCL) occurs in 2–10% of patients over their lifetime. Presents with rapidly enlarging lymph nodes, rising LDH, new B symptoms, and extranodal involvement. Prognosis is poor, with median survival of 5–8 months; treatment with R-CHOP or clinical trial enrolment is recommended.
- Secondary malignancies: CLL patients have a 2–3-fold increased risk of secondary cancers, including skin cancers (melanoma, squamous cell carcinoma), lung cancer, and therapy-related myelodysplastic syndrome (MDS)/acute myeloid leukaemia (AML).
- Cytopenias from marrow infiltration: Progressive bone marrow failure with anaemia and thrombocytopenia — may necessitate red cell or platelet transfusion support.
- Bone marrow failure: Progressive marrow infiltration by CLL cells leads to anaemia, thrombocytopenia, and neutropenia, necessitating transfusion support and acting as a treatment indication.
Investigations (Immunophenotyping)
Diagnostic Workup
Diagnosis of CLL requires a clonal B-cell population ≥5 × 10⁹/L in peripheral blood, persisting for ≥3 months, with characteristic immunophenotype on flow cytometry. A tissue biopsy is not required for diagnosis in most cases.
Essential Baseline Investigations
Prognostic & Staging Investigations
Risk Stratification & Prognostic Scoring
Modern CLL prognostication integrates clinical staging (Rai/Binet) with molecular and cytogenetic features to guide treatment timing and regimen selection. The CLL International Prognostic Index (CLL-IPI) combines five variables into a validated scoring system:
| CLL-IPI Factor | Points |
|---|---|
| TP53 disruption (del(17p) and/or TP53 mutation) | 4 |
| IGHV unmutated status | 2 |
| Serum β₂-microglobulin >3.5 mg/L | 2 |
| Clinical stage Binet B/C or Rai I–IV | 1 |
| Age >65 years | 1 |
Treatment Indications (iwCLL 2018 Criteria)
Treatment is not based on lymphocyte count alone. Therapy is indicated when any of the following are present:
- Evidence of progressive marrow failure: worsening anaemia (Hb <100 g/L) or thrombocytopenia (platelets <100 × 10⁹/L).
- Massive (≥6 cm below costal margin), progressive, or symptomatic splenomegaly.
- Massive (≥10 cm), progressive, or symptomatic lymphadenopathy.
- Progressive lymphocytosis with an increase of >50% over 2-month period or lymphocyte doubling time (LDT) <6 months (in the absence of autoimmune cytopenias).
- Autoimmune anaemia and/or thrombocytopenia refractory to corticosteroids or standard therapy.
- Constitutional symptoms: ≥10% weight loss over 6 months, significant fatigue (ECOG ≥2), fever >38°C for ≥2 weeks without infection, drenching night sweats for >1 month.
Management: Watch-and-Wait, Chemoimmunotherapy & Targeted Agents
Watch-and-Wait (Active Surveillance)
For patients with early-stage disease (Rai 0, Binet A) and no treatment indications, active surveillance with regular clinical and laboratory monitoring is standard of care. Multiple randomised trials have demonstrated no survival benefit from early treatment of asymptomatic CLL.
First-Line Treatment Selection
Treatment selection is guided by patient fitness, age, comorbidities (Cumulative Illness Rating Scale — CIRS), renal function, and — critically — the presence or absence of del(17p)/TP53 mutation.
Chemoimmunotherapy Regimens
Targeted Therapies (PBS-Listed in Australia)
Second-Line & Relapsed/Refractory Therapy
Treatment of relapsed/refractory CLL depends on the nature of prior therapy, duration of remission, and the presence of TP53 aberration. General principles:
- After FCR with long remission (≥3 years, mutated IGHV): Re-treatment with FCR or BR is reasonable if no TP53 aberration.
- After FCR with short remission (<3 years) or TP53 aberration: Switch to a non-chemotherapy regimen — ibrutinib, zanubrutinib, or venetoclax + rituximab.
- After BTK inhibitor failure: Venetoclax-based regimen (venetoclax + rituximab — 24 months); consider CAR-T cell therapy or bispecific antibodies in clinical trials.
- After venetoclax failure: BTK inhibitor if not previously used; consider pirtobrutinib (non-covalent BTK inhibitor) or clinical trial enrolment.
| Clinical Scenario | Preferred First-Line | Alternative |
|---|---|---|
| Fit, young, mutated IGHV, no TP53 aberration | FCR × 6 | Venetoclax + obinutuzumab (fixed 12 months) |
| Elderly or unfit (CIRS >6, eGFR <70) | Venetoclax + obinutuzumab (fixed 12 months) | Ibrutinib or zanubrutinib (continuous) |
| del(17p) or TP53 mutation (any age/fitness) | Ibrutinib or zanubrutinib (continuous) | Venetoclax + obinutuzumab (fixed 12 months) |
| Relapsed/refractory after chemoimmunotherapy | Ibrutinib or venetoclax + rituximab | Zanubrutinib; clinical trial |
Monitoring
During Watch-and-Wait
- FBC with differential every 3–6 months.
- Clinical examination (lymph nodes, spleen) at each visit.
- Immunoglobulin levels annually (guide IVIg replacement if IgG <4–5 g/L with recurrent infections).
- LDH and clinical assessment if rapid lymph node enlargement or new B symptoms develop — evaluate for Richter transformation.
- Skin cancer screening annually (increased risk of secondary malignancies).
During Treatment
- FCR/BR: FBC prior to each cycle (day 1); renal and hepatic function; monitor for infections. CMV reactivation surveillance if indicated.
- Ibrutinib/Zanubrutinib: FBC monthly for first 3 months, then 3-monthly. ECG at baseline and if symptoms of arrhythmia (palpitations). Monitor blood pressure regularly. Renal function 3–6 monthly. Assess for bleeding and drug interactions (CYP3A4 inhibitors — avoid ketoconazole, clarithromycin, grapefruit).
- Venetoclax: Electrolytes, creatinine, uric acid, phosphate, potassium at each dose escalation step (especially 20 mg, 50 mg). FBC every 2–4 weeks during ramp-up, then monthly. Assess MRD (flow cytometry on peripheral blood) at 12 months to evaluate depth of remission.
Minimal Residual Disease (MRD)
MRD assessment by high-sensitivity flow cytometry (sensitivity 10⁻⁴) is increasingly used to evaluate response depth, particularly after fixed-duration venetoclax-based therapy. MRD negativity (undetectable MRD) at the end of treatment is associated with prolonged progression-free survival. MRD-guided treatment strategies are under investigation in clinical trials and may inform future Australian practice.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760.
- 2. Eichhorst B, Fink AM, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17(7):928–942.
- 3. Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380(23):2225–2236. (CLL14 study)
- 4. Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–443. (E1912 study)
- 5. Tam CS, Brown JR, Kahl BS, et al. Zanubrutinib versus bendamustine plus rituximab in untreated chronic lymphocytic leukaemia and small lymphocytic lymphoma (SEQUOIA): a randomised, controlled, phase 3 study. Lancet Oncol. 2022;23(8):1031–1043.
- 6. International CLL-IPI Working Group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol. 2016;17(6):779–790.
- 7. Australian Institute of Health and Welfare (AIHW). Cancer in Australia 2023. AIHW, Canberra. Available from: https://www.aihw.gov.au/reports/cancer/cancer-in-australia
- 8. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17(6):768–778.
- 9. Thompson PA, Tam CS, O'Brien SM, et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term durability in a subset of patients with previously untreated CLL. J Clin Oncol. 2016;34(16):1888–1895.
- 10. Sahu KK, Sher T, Chan A, et al. Richter transformation: a review of clinical, pathological, and molecular features. Blood Rev. 2022;54:100930.
- 11. Australian Government Department of Health. Pharmaceutical Benefits Scheme — Venetoclax (PBS Item 12059). Available from: https://www.pbs.gov.au/
- 12. Hallek M, Shanafelt TD, Eichhorst B. Chronic lymphocytic leukaemia. Lancet. 2018;391(10129):1524–1537.
- 13. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax–rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107–1120. (MURANO study)
- 14. Royal Australasian College of Physicians. Adult Haematology Advanced Training Curriculum. Sydney: RACP; 2023.
- 15. Palliative Care Australia. National Palliative Care Standards. 5th ed. Canberra: PCA; 2018.