📋 Key Information Summary
- Pulmonary arterial hypertension (PAH) in connective tissue disease (CTD) is classified as WHO Group 1; systemic sclerosis (SSc) carries the highest risk with a 10–15% lifetime prevalence of SSc-PAH.
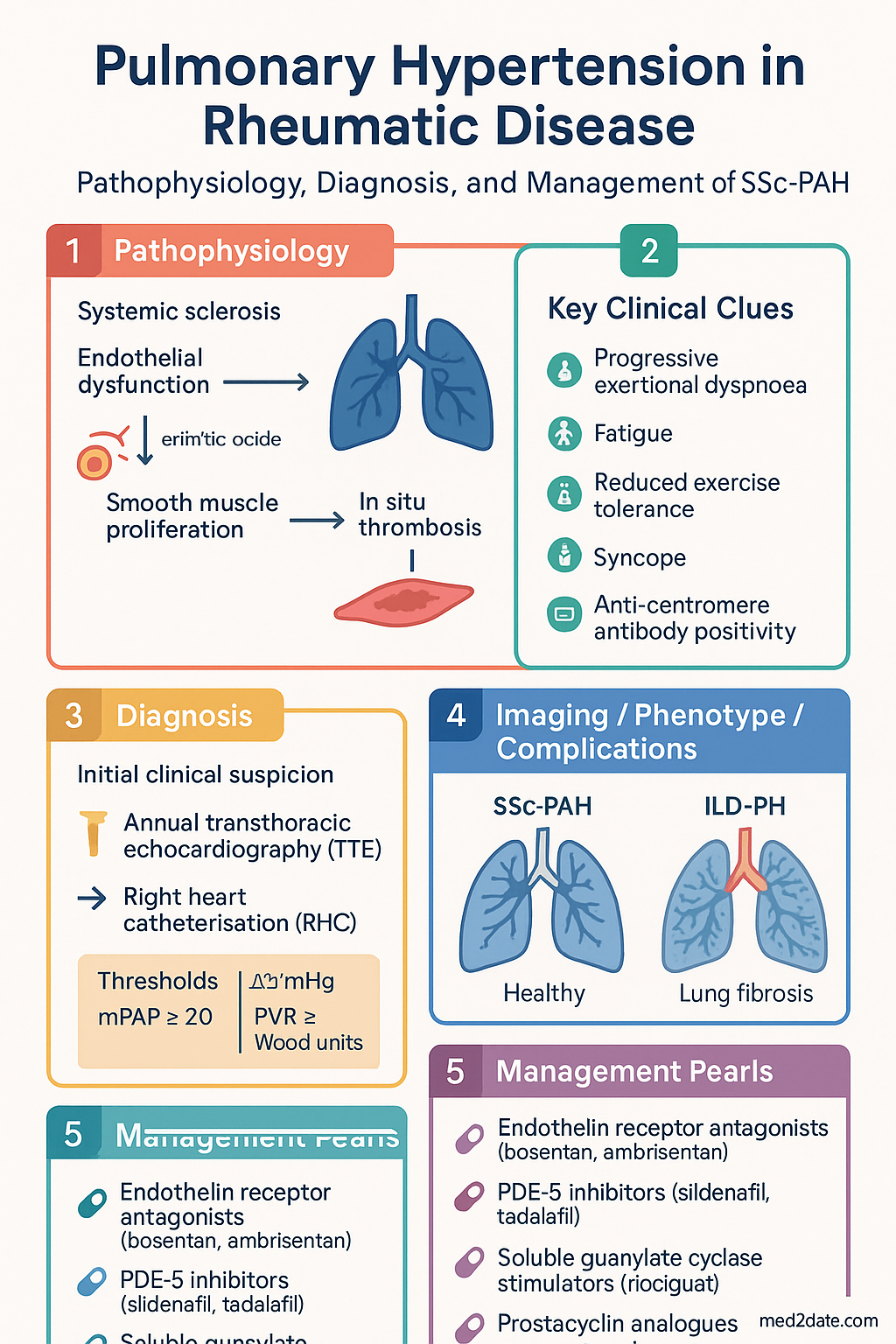
- Annual transthoracic echocardiography (TTE) screening is mandatory for all SSc patients from diagnosis — earlier screening is recommended if new symptoms arise.
- Right heart catheterisation (RHC) remains the gold standard for confirmation: mean pulmonary artery pressure (mPAP) ≥ 20 mmHg and pulmonary vascular resistance (PVR) ≥ 2 Wood units (WU).
- The DETECT algorithm (2012) provides a validated screening pathway for SSc patients to identify those requiring RHC referral.
- Differentiate WHO Group 1 PAH from Group 2 (left-heart disease) and Group 3 (ILD-associated PH) — treatment strategies differ substantially.
- Anti-centromere antibody positivity and limited cutaneous SSc are associated with a higher risk of isolated PAH; diffuse SSc with anti-Scl-70 increases ILD-PH risk.
- Low DLCO on pulmonary function testing (PFTs) is a key early marker — a DLCO < 60% predicted in a patient without significant fibrosis should prompt investigation for PAH.
- Targeted PAH therapies include endothelin receptor antagonists (bosentan, ambrisentan, macitentan), PDE-5 inhibitors (sildenafil, tadalafil), soluble guanylate cyclase stimulators (riociguat), and prostacyclin analogues (epoprostenol, iloprost, selexipag).
- Upfront combination therapy is recommended for intermediate- and high-risk patients per current ESC/ERS and Australian practice.
- BNP and NT-proBNP are essential prognostic markers; serial measurement guides treatment escalation.
- WHO functional class (I–IV) is the strongest single predictor of prognosis and guides treatment intensity.
- Referral for lung transplantation should be considered early for patients who fail to respond to optimised combination therapy or present in WHO FC III–IV.
- Aboriginal and Torres Strait Islander peoples with SSc face delayed diagnosis due to limited access to specialist screening and echocardiography services in remote areas.
- SSc-associated digital vasculopathy and renal crisis must be managed in parallel with PAH therapy; bosentan may reduce digital ulcer recurrence.
- SSc-PAH carries a guarded prognosis (median survival ~3 years without treatment) but targeted therapy has substantially improved outcomes.
Introduction & Australian Epidemiology
Pulmonary hypertension (PH) is a serious and potentially life-threatening complication of connective tissue diseases (CTDs), particularly systemic sclerosis (SSc). It is characterised by progressive remodelling of the pulmonary vasculature, leading to elevated pulmonary artery pressure, right ventricular failure, and death if untreated. Early detection through systematic screening and institution of targeted vasodilator therapy are the cornerstones of improved patient outcomes.
In Australia, SSc-associated PAH (SSc-PAH) is the most common form of CTD-PAH. The Australian Scleroderma Cohort Study (ASCS) has demonstrated a PAH prevalence of approximately 12% among SSc patients, with the limited cutaneous subset and anti-centromere antibody positivity carrying the greatest risk. Other CTDs associated with PAH include systemic lupus erythematosus (SLE), mixed connective tissue disease (MCTD), Sjögren syndrome, and inflammatory myopathies, though these are substantially less common.
PH in CTD can arise from multiple pathophysiological mechanisms classified by the WHO clinical grouping system. Differentiating WHO Group 1 PAH (pulmonary arterial hypertension) from Group 2 (left-heart disease) and Group 3 (lung disease–associated PH) is critical because treatment approaches diverge. SSc patients may harbour overlapping mechanisms, making accurate classification essential before commencing targeted therapy.
Australian data from the AIHW and the Australasian Society of Clinical Immunology and Allergy (ASCIA) registries indicate that the median age at SSc-PAH diagnosis is approximately 60 years, with a female predominance (3–4 : 1). Mortality remains significant, though the introduction of combination targeted therapy over the past two decades has reduced 3-year mortality from approximately 50% to under 30% in optimally treated cohorts.
Pathophysiology & Classification
WHO Classification of Pulmonary Hypertension
The Dana Point/Venice classification (updated at the 6th World Symposium, Nice 2018) categorises PH into five groups. CTD-related PH most commonly falls into Group 1 (PAH), but left-heart disease (Group 2) and interstitial lung disease (Group 3) are frequent confounders in the rheumatic disease population.
| WHO Group | Mechanism | CTD Relevance | Targeted PAH Therapy |
|---|---|---|---|
| Group 1 — PAH | Pulmonary arteriopathy; endothelial dysfunction, smooth muscle proliferation, in situ thrombosis; plexiform lesions | SSc (limited > diffuse), SLE, MCTD, Sjögren, PM/DM | Yes — primary indication |
| Group 2 — Left-heart disease | Passive back-pressure from diastolic or systolic LV dysfunction, valvular disease | SSc cardiac involvement, SLE myocarditis | No — treat underlying heart disease |
| Group 3 — Lung disease | Hypoxic vasoconstriction, vascular bed destruction from fibrosis | SSc-ILD (anti-Scl-70), antisynthetase syndrome, SLE-ILD | Generally not (except select combined cases) |
| Group 4 — CTEPH | Chronic organised thromboembolic obstruction | Rare in CTD; antiphospholipid syndrome association | Surgical PEA; riociguat for inoperable |
| Group 5 — Unclear/multifactorial | Haematological, metabolic, miscellaneous | Sarcoidosis, chronic haemolytic anaemia | Case-by-case |
Pulmonary Vasculopathy in SSc
SSc-PAH is driven by a triad of vascular pathology:
- Endothelial dysfunction: Excess endothelin-1 (ET-1) secretion, reduced nitric oxide (NO) and prostacyclin production. This underpins the rationale for ERA and PDE-5i therapy.
- Smooth muscle and fibroblast proliferation: Intimal hyperplasia and medial hypertrophy of pulmonary arterioles, narrowing the vascular lumen.
- In situ thrombosis: A procoagulant milieu within the pulmonary microvasculature.
Unlike idiopathic PAH (IPAH), SSc-PAH typically shows less plexiform lesion formation and more diffuse distal arteriolar disease. The right ventricle adapts poorly to the increased afterload, and right ventricular (RV) failure is the usual terminal event.
Differentiating SSc-PAH from ILD-PH
A critical distinction: patients with extensive SSc-associated ILD (forced vital capacity [FVC] < 70% predicted, fibrosis on HRCT) may develop PH secondary to lung destruction (WHO Group 3). In mixed PH-ILD, treatment with targeted PAH therapy is controversial and should involve multidisciplinary specialist discussion. Isolated PAH (Group 1) in SSc typically shows relatively preserved lung volumes with disproportionately reduced DLCO.
WHO Functional Classification
Diagnosis & Screening
Screening Strategy in SSc
Given the insidious onset and poor prognosis of SSc-PAH, systematic screening is essential. The Australian Scleroderma Interest Group and international guidelines recommend:
- Annual transthoracic echocardiography (TTE) for all SSc patients from the time of diagnosis.
- Annual PFTs including DLCO — a falling DLCO disproportionate to FVC decline is a red flag.
- Symptom reassessment at every visit — progressive exertional dyspnoea, fatigue, reduced exercise tolerance, or syncope warrant urgent evaluation.
- Biomarkers: BNP or NT-proBNP at baseline and annually.
DETECT Algorithm (for SSc)
The DETECT algorithm (Coghlan et al., Ann Rheum Dis 2014) is a validated two-step screening tool for SSc patients with disease duration ≥ 3 years and DLCO < 60% predicted. Step 1 uses clinical and simple laboratory variables to generate a risk score; Step 2 adds echocardiographic and BNP data to determine the need for RHC referral.
Transthoracic Echocardiography (TTE) Criteria
TTE estimates the pulmonary artery systolic pressure (PASP) from the tricuspid regurgitation velocity (TRV) using the modified Bernoulli equation plus estimated right atrial pressure. Key thresholds:
- PASP > 40 mmHg or TRV > 3.0 m/s — suggestive of PH, warrants RHC.
- RV dilation, hypertrophy, or dysfunction on echo — increases pre-test probability.
- Pericardial effusion — associated with worse prognosis and higher likelihood of PAH.
Right Heart Catheterisation (RHC) — Gold Standard
RHC is mandatory to confirm PAH and classify the haemodynamic severity. The haemodynamic definitions (updated 6th World Symposium, 2018):
| Parameter | Definition |
|---|---|
| Pre-capillary PH | mPAP ≥ 20 mmHg AND PAWP ≤ 15 mmHg AND PVR ≥ 2 WU |
| Isolated post-capillary PH | mPAP ≥ 20 mmHg AND PAWP > 15 mmHg AND PVR < 2 WU |
| Combined pre- and post-capillary PH | mPAP ≥ 20 mmHg AND PAWP > 15 mmHg AND PVR ≥ 2 WU |
In SSc, pure pre-capillary PAH (Group 1) should show PAWP ≤ 15 mmHg. A raised PAWP suggests left-heart involvement and should redirect treatment focus. Acute vasodilator testing with inhaled nitric oxide or IV epoprostenol should be performed at RHC to identify vasoreactive responders (rare in SSc-PAH but important when present).
Additional Investigations
Risk Stratification & Severity Scoring
Risk stratification at baseline and during follow-up determines treatment intensity. The ESC/ERS and REVEAL risk calculators integrate WHO FC, 6MWD, BNP/NT-proBNP, RHC haemodynamics, and echo parameters. In clinical practice, a simplified three-strata approach is commonly used:
Empirical & Initial Therapy
Supportive therapies should be initiated alongside targeted agents. In CTD-PAH, upfront combination therapy is now standard for patients at intermediate or high risk.
Supportive Measures
- Diuretics: Loop diuretics (furosemide 20–80 mg PO daily, or IV for acute decompensation) for volume overload and RV failure. Titrate to euvolaemia.
- Supplemental oxygen: Maintain SpO₂ ≥ 90% at rest and during exertion. MBS-subsidised home oxygen available via State/Territory programmes.
- Anticoagulation: Routine anticoagulation is not recommended for SSc-PAH due to bleeding risk (especially GI telangiectasia). Consider only if thromboembolic aetiology or immobilisation.
- Exercise rehabilitation: Supervised pulmonary rehabilitation improves 6MWD and quality of evidence; available at major centres.
- Pregnancy avoidance: ERA and PDE-5i are teratogenic — reliable contraception is mandatory. WHO FC III–IV carries extreme pregnancy risk.
Targeted PAH Therapies — Three Pathways
Current targeted therapies act on three molecular pathways involved in pulmonary vascular remodelling:
| Pathway | Drug Class | Australian-Approved Agents |
|---|---|---|
| Endothelin pathway | Endothelin receptor antagonists (ERAs) | Bosentan, ambrisentan, macitentan |
| NO–sGC–cGMP pathway | PDE-5 inhibitors; sGC stimulators | Sildenafil, tadalafil; riociguat |
| Prostacyclin pathway | Prostacyclin analogues; IP receptor agonists | Epoprostenol (IV), iloprost (inhaled), selexipag (oral) |
Directed / Pathogen-Specific Therapy — Drug Details
Endothelin Receptor Antagonists (ERAs)
PDE-5 Inhibitors
Soluble Guanylate Cyclase Stimulator
Prostacyclin Analogues & IP Receptor Agonists
Recommended Combination Regimens
Current ESC/ERS and Australian expert consensus recommend upfront combination therapy for intermediate- and high-risk patients:
| Risk Category | First-Line Combination | Escalation |
|---|---|---|
| Low risk (WHO FC I–II) | ERA + PDE-5i (e.g. ambrisentan + tadalafil) | Add selexipag or switch to triple therapy |
| Intermediate risk (WHO FC III) | Dual oral: ERA + PDE-5i | Add selexipag or inhaled iloprost; consider IV epoprostenol |
| High risk (WHO FC III–IV) | IV epoprostenol + dual oral therapy | Transplant listing if refractory |
Monitoring
Structured follow-up is essential for treatment-to-target management. Recommended monitoring schedule:
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2016;37(1):67-119.
- 2. Coghlan JG, Denton CP, Grünig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014;73(7):1340-1349.
- 3. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618-3731.
- 4. Proudman SM, Stevens WM, Sahhar J, Celermajer D. Pulmonary arterial hypertension in systemic sclerosis: the need for early detection and treatment. Intern Med J. 2007;37(7):485-494.
- 5. Nikpour M, Hissaria P, Byron J, Sahhar J, Micallef M, Pender M, et al. Prevalence, correlates and clinical usefulness of antibodies to RNA polymerase III in systemic sclerosis: a cross-sectional analysis of data from an Australian cohort. Arthritis Res Ther. 2011;13(6):R211.
- 6. Galie N, Barberà JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834-844 (AMBITION trial).
- 7. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809-818 (SERAPHIN trial).
- 8. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522-2533 (GRIPHON trial).
- 9. Ghofrani HA, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330-340 (PATENT-1 trial).
- 10. Australian Institute of Health and Welfare (AIHW). Scleroderma in Australia. Cat. no. PHE 292. Canberra: AIHW; 2021.
- 11. Launay D, Sobanski V, Hachulla E, Humbert M. Pulmonary hypertension in systemic sclerosis: different phenotypes. Eur Respir Rev. 2017;26(145):170056.
- 12. The Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice (Red Book), 9th edn. East Melbourne: RACGP; 2016 (updated 2018).
- 13. National Aboriginal Community Controlled Health Organisation (NACCHO). Aboriginal and Torres Strait Islander health performance framework: Tier 2 — Determinants of health. Canberra: Australian Government; 2020.