📋 Key Information Summary
- Adult-onset Still's disease (AOSD) is a rare systemic autoinflammatory disorder characterised by quotidian spiking fevers ≥39°C, an evanescent salmon-pink rash, arthritis, and markedly elevated serum ferritin.
- Diagnosis is clinical and requires exclusion of infection, malignancy (especially lymphoma), and other autoimmune rheumatic diseases; there is no single pathognomonic test.
- Yamaguchi criteria (≥5 features including ≥2 major) remain the standard diagnostic framework; major criteria: fever ≥39°C for ≥1 week, typical rash, arthralgia ≥2 weeks, leukocytosis with neutrophilia.
- Serum ferritin >3000 µg/L with glycosylated ferritin fraction <20% is highly specific for AOSD and should be requested as a first-line investigation.
- AOSD pathogenesis centres on an autoinflammatory cytokine storm driven by IL-1β, IL-18, and IL-6; this rationalises targeted biologic therapy.
- First-line therapy is NSAIDs for mild disease; systemic corticosteroids (prednisolone 0.5–1 mg/kg/day) remain the mainstay for moderate–severe presentations.
- Methotrexate is the preferred steroid-sparing agent; IL-1 inhibitors (anakinra, canakinumab) and IL-6 blockade (tocilizumab) are indicated for refractory or steroid-dependent disease.
- Macrophage activation syndrome (MAS) / secondary HLH is a life-threatening complication presenting with rapid cytopenias, hyperferritinaemia >10 000 µg/L, hepatitis, and coagulopathy; requires urgent high-dose methylprednisolone plus ciclosporin.
- Two clinical patterns are recognised: systemic (fever, serositis, rash, lymphadenopathy) and chronic articular (persistent polyarthritis resembling RA, but seronegative).
- Approximately 70% of patients achieve monocyclic or polycyclic remission; 20–30% develop chronic articular disease with risk of joint destruction requiring DMARD or biologic therapy.
- All serological markers (RF, ANA, anti-CCP) are characteristically negative; a positive RF or ANA should prompt reconsideration of the diagnosis.
- Aboriginal and Torres Strait Islander patients may present with delayed diagnosis due to barriers in specialist access; early referral to rheumatology and culturally safe care pathways are essential.
Introduction & Australian Epidemiology
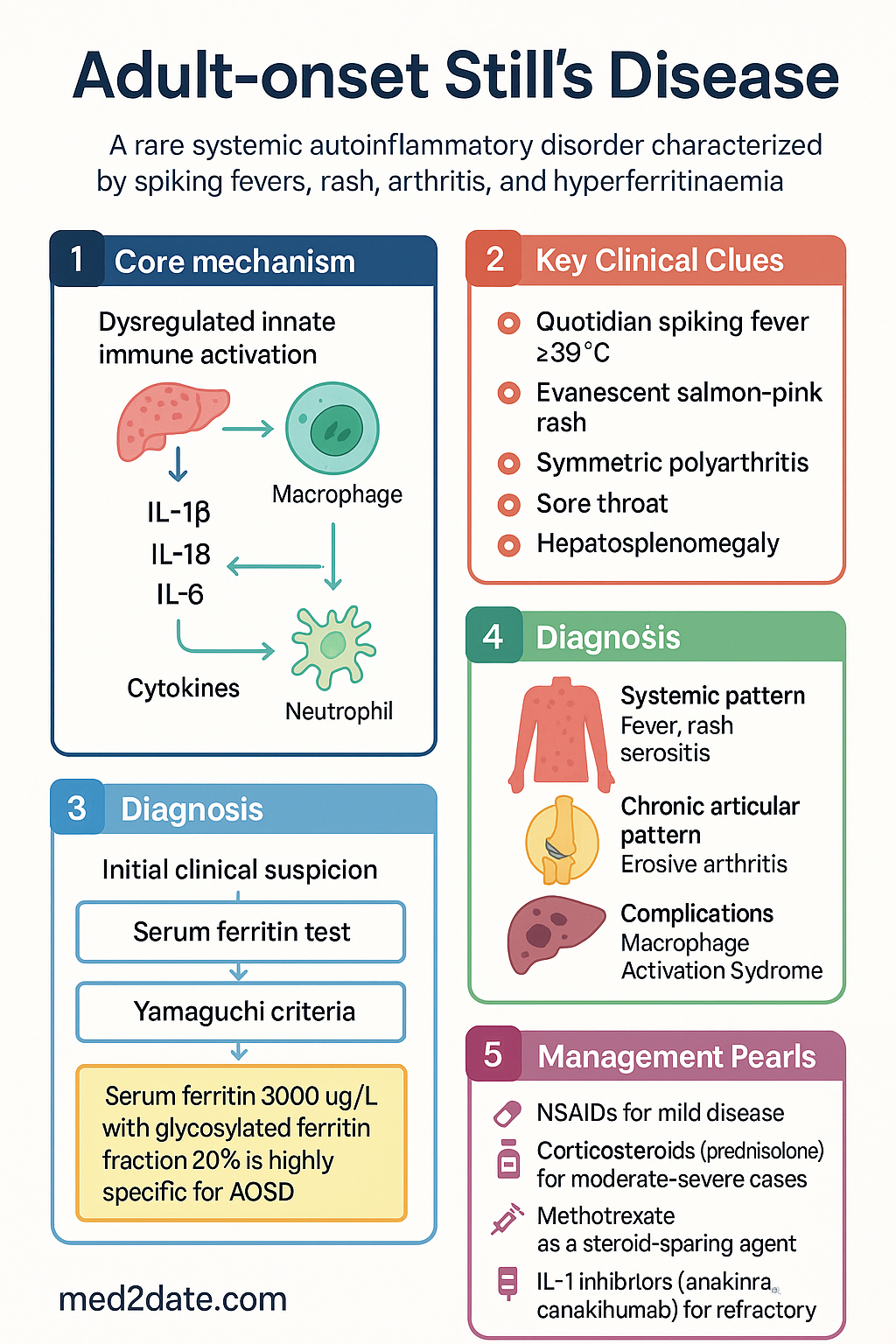
Adult-onset Still's disease (AOSD) is a rare systemic autoinflammatory disorder of unknown aetiology, considered the adult equivalent of systemic juvenile idiopathic arthritis (sJIA). The condition is characterised by quotidian (daily) spiking fevers, an evanescent salmon-pink macular rash, polyarthritis, and a pronounced acute-phase response dominated by extreme hyperferritinaemia. AOSD falls within the spectrum of autoinflammatory diseases driven by dysregulated innate immunity, particularly excessive interleukin-1β (IL-1β), IL-18, and IL-6 signalling.
AOSD is estimated to affect 0.1–0.4 per 100 000 population worldwide. Australian incidence data are limited, but tertiary rheumatology centres in Sydney, Melbourne, and Brisbane report managing 2–5 new cases annually per centre. The disease shows a bimodal age distribution with peaks at 15–25 years and 36–46 years, and no significant sex predilection in most series. Diagnosis is typically delayed by 3–6 months because the presentation mimics infection, lymphoma, and other systemic autoimmune conditions.
In Australia, clinicians should be aware of the high background prevalence of infections (Q fever, rickettsial disease, Ross River virus in endemic areas) that may present with fever, rash, and arthralgia — features overlapping with AOSD. A thorough infectious disease workup is mandatory before committing to an AOSD diagnosis.
Pathogenesis & Diagnostic Criteria
Pathogenesis
AOSD is classified among the monogenic and polygenic autoinflammatory diseases. The hallmark is dysregulated innate immune activation producing a cytokine storm without evidence of adaptive autoimmune T- or B-cell–mediated pathology (hence seronegative status). Key pathogenic mediators include:
- IL-1β: The dominant cytokine. Activated macrophages and neutrophils release IL-1β via the NLRP3 inflammasome, driving fever, rash, synovitis, and acute-phase reactant elevation. IL-1β blockade is the most effective targeted therapy.
- IL-18: Markedly elevated in AOSD (often >10 × upper limit of normal); correlates with disease activity and MAS risk. IL-18–binding protein levels may be relatively insufficient.
- IL-6: Produced downstream of IL-1β stimulation; responsible for CRP, ferritin, and hepcidin upregulation. Contributes to the anaemia of chronic disease.
- IL-17 and Th17 axis: Emerging evidence of Th17 skewing, particularly in the chronic articular phenotype.
- Neutrophil activation: Neutrophilia and neutrophilic infiltration of synovium, liver, serosal surfaces, and reticuloendothelial system explain the organomegaly and serositis.
Diagnostic Criteria
No single test confirms AOSD. The Yamaguchi criteria (1992) remain the most widely validated classification set. They require ≥5 features including ≥2 major criteria, with exclusion of infection, malignancy, and other rheumatic diseases.
| Yamaguchi Criterion | Major | Minor |
|---|---|---|
| Fever ≥39°C for ≥1 week | ✔ | |
| Typical rash (evanescent, salmon-pink, coinciding with fever) | ✔ | |
| Arthralgia or arthritis ≥2 weeks | ✔ | |
| Leukocytosis (≥10 × 10⁹/L) with neutrophilia (≥80%) | ✔ | |
| Sore throat | ✔ | |
| Lymphadenopathy and/or splenomegaly | ✔ | |
| Liver dysfunction (elevated transaminases) | ✔ | |
| Negative RF and ANA | ✔ |
Supportive Biomarkers (Not in Yamaguchi but Clinically Essential)
- Serum ferritin >1000 µg/L: Present in >80% of patients; values >3000 µg/L are highly suggestive.
- Glycosylated ferritin <20%: Highly specific for AOSD; normal glycosylated ferritin is 20–50%. Request this test early — available at major Australian hospital laboratories (MBS item 66835 for serum ferritin; glycosylated fraction may require specialist laboratory referral).
- Elevated IL-18: Research biomarker; not yet standard in Australian pathology panels but available at reference laboratories.
- Marked elevation of ESR, CRP, LDH: Non-specific but correlate with disease activity.
Clinical Features & Complications
Classic Presentation
AOSD typically presents with a dramatic systemic illness. The hallmark features are:
- Quotidian (daily) spiking fever: Temperature ≥39°C, often rising to 40°C in the late afternoon/evening, then returning to normal or subnormal by morning. This double-quotidian pattern (two spikes per day) is virtually pathognomonic when present.
- Salmon-pink evanescent rash: A faint macular or maculopapular rash, 2–5 mm, non-pruritic, classically on the trunk and proximal limbs, typically appearing during febrile episodes and fading within hours. Koebner phenomenon may occur. Skin biopsy shows mild perivascular lymphocytic infiltration.
- Arthritis: Usually symmetric polyarthritis affecting wrists, knees, ankles, and small joints of the hands. May present early as arthralgia without clinical synovitis. Chronic articular disease can cause erosive joint destruction resembling rheumatoid arthritis.
- Sore throat: Severe pharyngitis, often preceding or coinciding with febrile episodes; may be the presenting complaint.
- Hepatosplenomegaly: Mild–moderate hepatomegaly and/or splenomegaly due to reticuloendothelial activation.
- Lymphadenopathy: Generalised, non-tender; occasionally marked enough to raise lymphoma concern.
- Serositis: Pleuritis, pericarditis, or both; pericardial effusion may be clinically significant.
Two Clinical Phenotypes
| Feature | Systemic Pattern | Chronic Articular Pattern |
|---|---|---|
| Proportion | ~60–70% | ~30–40% |
| Fever | Dominant, quotidian | Less prominent or absent |
| Rash | Prominant, evanescent | Mild or absent |
| Arthritis | Mild, self-limiting | Deforming, erosive |
| Course | Monocyclic (~60%) or polycyclic (~15%) | Chronic, persistent (~25%) |
| Treatment | NSAIDs → steroids ± biologics | DMARDs + biologics |
Complications
HLH-2004 Diagnostic Criteria for MAS (Modified for AOSD)
- Fever
- Splenomegaly
- Cytopenias (2 of 3: Hb <90 g/L, platelets <100 × 10⁹/L, neutrophils <1.0 × 10⁹/L)
- Hypertriglyceridaemia (>2.0 mmol/L) and/or hypofibrinogenaemia (<1.5 g/L)
- Haemophagocytosis on bone marrow, spleen, or lymph node biopsy
- Hyperferritinaemia >10 000 µg/L (AOSD-adapted threshold)
- Elevated soluble IL-2 receptor (sCD25) >2400 U/mL
- Low or absent NK-cell activity
Other Complications
- Diffuse alveolar haemorrhage: Rare but life-threatening; presents with haemoptysis, hypoxia, and diffuse ground-glass opacities on CT chest.
- Myocarditis / pericarditis: May cause tamponade or heart failure.
- Reactive amyloidosis (AA): Chronic inflammation may lead to AA amyloidosis with nephrotic syndrome; monitors with urinary protein-to-creatinine ratio.
- Drug-related complications: Steroid osteoporosis, avascular necrosis, diabetes; methotrexate hepatotoxicity; biologic infection risk.
- Thrombotic microangiopathy: Reported in severe AOSD; may mimic TTP/HUS.
Investigations
Investigation in AOSD serves two purposes: supporting the diagnosis and excluding mimics. A systematic approach is essential.
Baseline Investigations
Exclusion Investigations
Imaging
- Synovial ultrasound/MRI: Detects early synovitis and erosions in chronic articular disease.
- Echocardiography: If pericarditis or myocarditis suspected; pericardial effusion is common.
- High-resolution CT chest: If pulmonary symptoms; ground-glass opacities may indicate DAH or organising pneumonia.
Risk Stratification & Disease Course
AOSD follows three recognised clinical courses, each with distinct prognostic implications:
Predictors of Poor Outcome
- Chronic articular disease pattern
- Steroid dependence / refractory disease
- Development of MAS
- Hip or large-joint arthritis at presentation
- Elevated IL-18 and ferritin at baseline (paradoxically associated with worse outcomes)
- Delay to diagnosis >6 months
Management
Treatment of AOSD follows a stepwise approach from NSAIDs to corticosteroids, conventional DMARDs, and targeted biologics. The choice depends on disease severity, pattern (systemic vs chronic articular), and response to therapy.
Stepwise Treatment Algorithm
Drug Detail
Management of MAS / HLH
- Immediate: IV methylprednisolone 500–1000 mg daily × 3 days
- Concurrent: Ciclosporin 2–5 mg/kg/day (oral or IV) — initiate within 24 hours of MAS diagnosis
- If refractory to 48 hours of high-dose steroids + ciclosporin: Consider anakinra 1–2 mg/kg/day SC/IV; etoposide (per HLH-94 protocol) only under haematology guidance
- Supportive: ICU admission for haemodynamic monitoring; blood product support (FFP, cryoprecipitate for DIC); avoid unnecessary NSAIDs (renal/bleeding risk)
- Treat trigger: If concurrent infection identified, treat aggressively; stop any causative drug
Treatment of Chronic Articular Disease
The chronic articular phenotype requires a rheumatoid arthritis–like approach:
- Methotrexate: First-line DMARD; escalate to 25 mg/week if needed.
- Leflunomide: Alternative if MTX contraindicated; 20 mg/day PO.
- Biologics: IL-1 inhibitors (anakinra) for systemic flares; tocilizumab or TNF inhibitors (adalimumab, etanercept) for articular predominance.
- Intra-articular corticosteroids: For persistent mono/oligoarthritis.
Monitoring
Disease Activity Monitoring
- CRP and ESR: Every 2–4 weeks during active treatment; every 1–3 months in remission.
- Serum ferritin and glycosylated ferritin: Baseline and with each flare; falling ferritin and rising glycosylated fraction indicates response.
- FBC: Monitor for cytopenias (MAS) and drug toxicity (MTX, ciclosporin).
- LFTs:
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still's disease. Autoimmun Rev. 2014;13(7):708-722.
- 2. Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19(3):424-430.
- 3. Fautrel B, Zing E, Golmard JL, et al. Proposal for a new set of classification criteria for adult-onset Still disease. Medicine (Baltimore). 2002;81(3):194-200.
- 4. Mitrovic S, Fautrel B. New insights into the pathogenesis and the therapeutic landscape of adult-onset Still's disease. Joint Bone Spine. 2018;85(5):547-553.
- 5. Hinojosa-Azaola A, García-García C, Jiménez-Balderas FJ. Macrophage activation syndrome in adult-onset Still's disease. Med Clin (Barc). 2018;151(8):323-329.
- 6. Colafrancesco S, Priori R, Valesini G, et al. Response to interleukin-1 inhibitors in adult-onset Still's disease: a nationwide study of 103 Italian patients. J Rheumatol. 2019;46(12):1647-1652.
- 7. Nirmala N, Brachat A, Feist E, et al. Gene-expression analysis of adult-onset Still's disease and systemic juvenile idiopathic arthritis is consistent with a continuum of a single disease entity. Pediatr Rheumatol. 2015;13:50.
- 8. Helliwell T, Muller S, Hider SL, et al. Challenges of diagnosing and managing adult-onset Still's disease in primary care. Rheumatol Adv Pract. 2020;4(2):rkaa043.
- 9. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023. Canberra: AIHW; 2023.
- 10. Ranzolin A, Bredemeier M, Wobeto C, et al. Adult-onset Still's disease in South America: a Brazilian multicentre study. Clin Rheumatol. 2020;39(7):2129-2136.
- 11. Efthimiou P, Kontzias A, Ward CM, Ogden NS. Adult-onset Still's disease: can recent advances in our understanding of its pathogenesis lead to targeted therapy? Nat Clin Pract Rheumatol. 2007;3(6):328-335.
- 12. Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140-1146.
- 13. National Health and Medical Research Council (NHMRC). Australian Guidelines for the Clinical Care of People with CKD. Canberra: NHMRC; 2024 (relevant for renal dosing guidance).
- 14. Ruscitti P, Cipriani P, Masedu F, et al. Adult-onset Still's disease: evaluation of prognostic tools and validation of the systemic score by analysis of 100 cases from three centers. BMC Med. 2016;14:194.
- 15. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.