📋 Key Information Summary
- Sarcoidosis is a multisystem granulomatous disorder of unknown aetiology characterised by non-caseating granulomas in affected organs
- Pulmonary involvement occurs in >90% of cases; bilateral hilar lymphadenopathy with or without pulmonary infiltrates is the most common presentation
- Lofgren syndrome — erythema nodosum + bilateral hilar lymphadenopathy + polyarthralgia — carries an excellent prognosis with >90% spontaneous resolution
- Serum ACE level supports diagnosis but lacks sensitivity and specificity; elevated in ~60% of active sarcoidosis and can be raised in granulomatous infections
- Tissue biopsy demonstrating non-caseating granulomas with negative AFB/fungal stains is the gold standard for diagnosis
- Cardiac sarcoidosis can present with arrhythmias, heart block, or sudden cardiac death; cardiac MRI and PET-CT are key investigations
- Neurosarcoidosis affects cranial nerves (especially CN VII) and can cause hypothalamic/pituitary dysfunction including central diabetes insipidus
- Oral corticosteroids (prednisolone 20–40 mg daily) remain first-line therapy for symptomatic disease requiring treatment
- Methotrexate (10–15 mg/week) is the preferred steroid-sparing agent; azathioprine and mycophenolate are alternatives
- Infliximab (PBS Authority Required for sarcoidosis) is reserved for refractory disease or when conventional steroid-sparing agents fail
- Aboriginal and Torres Strait Islander peoples may have higher rates of sarcoidosis-related morbidity with reduced access to specialist rheumatology services in remote regions
- Monitoring includes pulmonary function tests, serum ACE, serum calcium, serum creatinine, ECG, and ophthalmological assessment at baseline and follow-up
Introduction & Australian Epidemiology
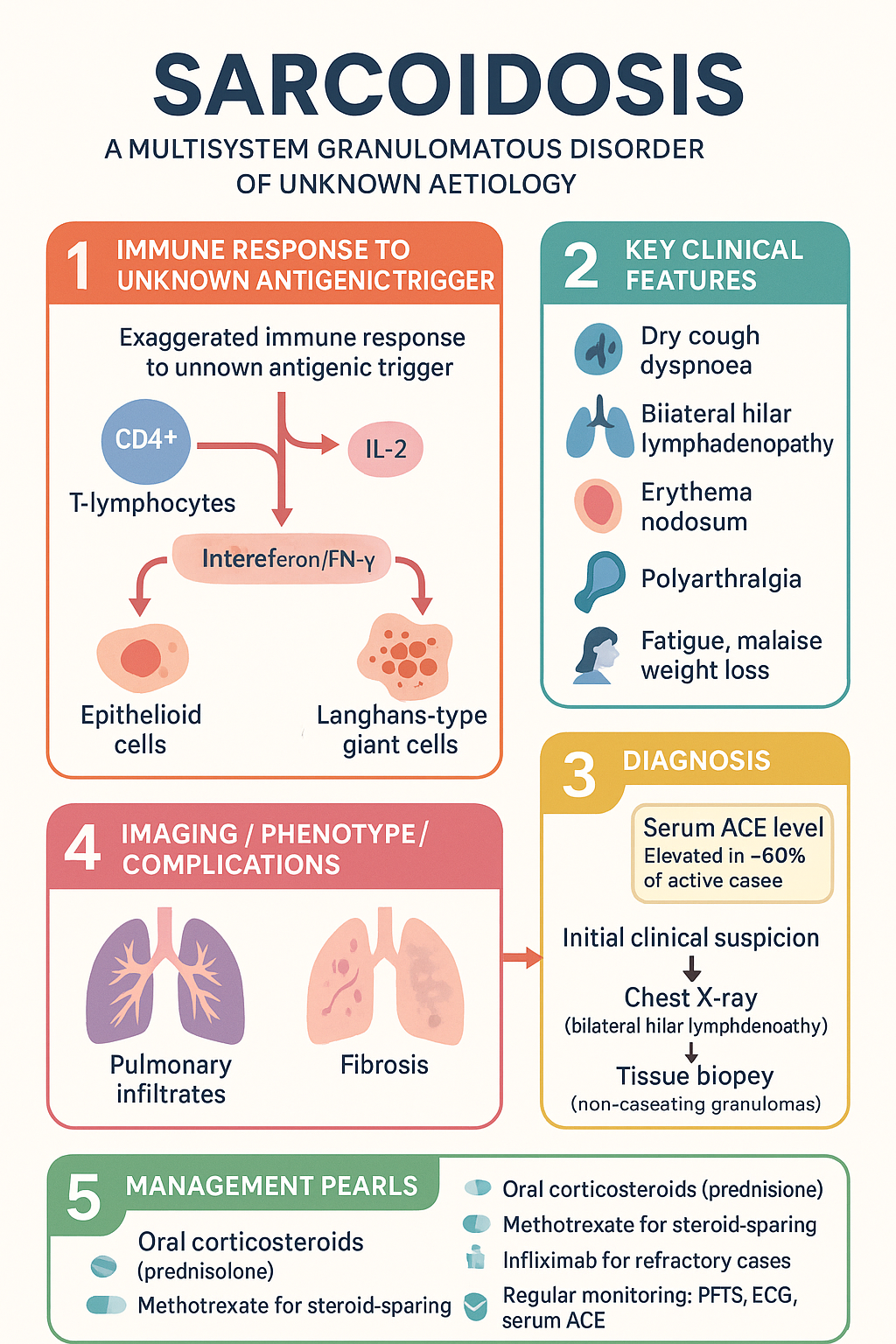
Sarcoidosis is a systemic granulomatous disorder of unknown aetiology that predominantly affects the lungs, lymph nodes, skin, eyes, and heart. The condition is characterised by the formation of non-caseating granulomas — compact clusters of activated macrophages and T-helper cells — in one or more organs. The hallmark histological finding must be distinguished from other granulomatous diseases, including tuberculosis, fungal infections, and berylliosis.
In Australia, sarcoidosis has an estimated incidence of 5–10 per 100,000 population, with higher rates reported among individuals of Northern European and Scandinavian descent. The disease has a bimodal age distribution with peaks between 25–35 years and 50–65 years. There is a slight female predominance. Compared to non-Indigenous Australians, Aboriginal and Torres Strait Islander peoples may present with more advanced disease at diagnosis, compounded by geographic barriers to specialist care.
The aetiology remains elusive but is thought to involve an exaggerated immune response to an unknown antigenic trigger in genetically predisposed individuals. Environmental exposures to moulds, organic dusts, insecticides, and metalworking fluids have been implicated. Genetic susceptibility is linked to HLA-DRB1 alleles and polymorphisms in the BTNL2 gene, among others.
Approximately 50–60% of patients with sarcoidosis require no treatment and experience spontaneous remission within 2–3 years. However, 10–30% develop chronic disease with progressive pulmonary fibrosis, cardiac involvement, or other organ damage leading to significant morbidity. Overall mortality is approximately 1–5%, primarily attributed to pulmonary, cardiac, or neurological complications.
Pulmonary Sarcoidosis
The lungs are the most commonly affected organ in sarcoidosis, with involvement in over 90% of cases. Pulmonary sarcoidosis ranges from asymptomatic bilateral hilar lymphadenopathy to progressive fibrotic lung disease. Understanding the radiographic staging system guides prognosis and management decisions.
Chest X-Ray Staging (Scadding Classification)
| Stage | Radiographic Findings | Spontaneous Resolution |
|---|---|---|
| Stage 0 | Normal chest X-ray | N/A — extrathoracic disease |
| Stage I | Bilateral hilar lymphadenopathy (BHL) alone | 60–90% |
| Stage II | BHL + pulmonary infiltrates | 40–70% |
| Stage III | Pulmonary infiltrates without BHL | 10–20% |
| Stage IV | Pulmonary fibrosis with volume loss, honeycombing | <5% |
Clinical Features
- Dry cough (most common), dyspnoea on exertion, chest discomfort
- Many patients with Stage I disease are asymptomatic — detected incidentally on CXR
- Wheeze is uncommon; crackles may be auscultated in advanced disease
- Constitutional symptoms: fatigue (predominant symptom in many series), malaise, weight loss, night sweats
- Progressive disease may lead to pulmonary hypertension from fibrotic parenchymal destruction
Pulmonary Function Testing
Spirometry typically demonstrates a restrictive pattern (reduced FVC, reduced FEV₁, preserved or elevated FEV₁/FVC ratio) reflecting granulomatous infiltration of the lung parenchyma. The DLCO (diffusing capacity) is reduced. In advanced disease with pulmonary hypertension, a mixed obstructive-restrictive pattern may emerge. Serial PFTs every 3–6 months guide treatment response.
Lofgren Syndrome
Lofgren syndrome is a specific acute presentation of sarcoidosis characterised by a classical triad: bilateral hilar lymphadenopathy, erythema nodosum, and polyarthralgia (especially ankle arthritis). It carries an excellent prognosis with spontaneous resolution in over 90% of cases.
Diagnostic Features
- Erythema nodosum: Bilateral, tender, erythematous nodules typically on the shins; most common panniculitis pattern in sarcoidosis
- Polyarthralgia: Predominantly affecting ankles, also knees and wrists; may be associated with periarticular swelling and tenosynovitis
- Bilateral hilar lymphadenopathy: Stage I radiographic appearance
- Fever is often present at presentation
- Elevated ESR and CRP; serum ACE may be normal in the acute presentation
Associated Features
- Female predominance (F:M ratio approximately 3:1 in Lofgren syndrome, reversing the usual slight female predominance of sarcoidosis overall)
- HLA-DRB1*03 association is strong, supporting a genetic basis
- Bilateral ankle periarthritis is characteristic and may be the presenting complaint
Management
Lofgren syndrome is typically self-limiting. Treatment is symptomatic with NSAIDs (naproxen 500 mg BD or ibuprofen 400 mg TDS) for joint pain and erythema nodosum. A short course of oral prednisolone (20–30 mg daily, tapering over 4–6 weeks) may be required for severe joint symptoms not responding to NSAIDs. Prognosis is excellent; recurrence is rare. Formal tissue biopsy is not always required when the clinical triad is classical.
Cutaneous Sarcoidosis
Skin involvement occurs in approximately 25–35% of patients with sarcoidosis and may be the presenting feature. Cutaneous lesions are useful as they are readily accessible for biopsy, providing histological confirmation of non-caseating granulomas. Cutaneous sarcoidosis is classified as specific (granulomas present on histology) or non-specific (reactive patterns without granulomas, e.g., erythema nodosum).
Specific Cutaneous Lesions
- Plaques: Annular, violaceous, infiltrated plaques — most common specific lesion type in Australia
- Papules: Small, translucent, brownish-red papules — perioral, perinasal, and periocular distribution
- Lupus pernio: Violaceous, indurated plaques and nodules on the nose, cheeks, ears, and digits — strongly associated with upper respiratory tract and sarcoid granulomas; often disfiguring
- Subcutaneous nodules: Firm, painless, deep dermal/subcutaneous nodules — Darier-Roussy variant
- Scar sarcoidosis: Infiltration of old scars with granulomatous tissue — pathognomonic when present
- Hypopigmented patches: More common in patients with darker skin phototypes
Non-Specific Cutaneous Lesions
- Erythema nodosum: The most common cutaneous finding in sarcoidosis — seen in Lofgren syndrome; represents a reactive panniculitis without granulomas
Management of Cutaneous Sarcoidosis
Localized cutaneous sarcoidosis may respond to potent topical corticosteroids (e.g., clobetasol propionate 0.05% applied BD for 2–4 weeks under occlusion for thick plaques). Intralesional triamcinolone (5–10 mg/mL) is effective for individual nodules and plaques. For widespread or disfiguring disease, systemic therapy with oral prednisolone, methotrexate, or hydroxychloroquine (200 mg BD) may be required. Lupus pernio is particularly refractory and often requires systemic agents. Referral to dermatology for consideration of laser therapy or biologic agents may be appropriate for cosmetic concern.
Cardiac & Neurosarcoidosis
Cardiac Sarcoidosis
Cardiac involvement is found in 20–30% of sarcoidosis patients at autopsy, although clinically apparent disease is reported in approximately 5% of cases. Cardiac sarcoidosis is a major cause of sarcoidosis-related mortality, accounting for up to 50–65% of deaths in some series.
Clinical Presentations
- Conduction abnormalities: AV block (first-, second-, or third-degree), bundle branch block, intraventricular conduction delay — most common presentation
- Arrhythmias: Ventricular tachycardia (VT), atrial fibrillation, atrial flutter — VT from granulomatous myocardial scarring is the primary cause of sudden cardiac death
- Heart failure: Systolic or diastolic dysfunction from myocardial granulomatous infiltration or fibrotic cardiomyopathy
- Pericarditis: Pericardial effusions, rarely constrictive pericarditis
- Sudden cardiac death: May be the first manifestation of cardiac sarcoidosis
Diagnostic Approach
Neurosarcoidosis
Neurological involvement occurs in 5–15% of patients with sarcoidosis. The central and peripheral nervous systems may be affected simultaneously or independently. Neurosarcoidosis can mimic many neurological conditions and is often a diagnosis of exclusion.
Clinical Patterns
- Cranial neuropathies: Facial nerve palsy (CN VII — most common, ~50% of neurosarcoidosis), optic neuritis (CN II), vestibulocochlear dysfunction (CN VIII)
- Hypothalamic–pituitary involvement: Central diabetes insipidus, hyperprolactinaemia, hypopituitarism, temperature dysregulation
- Meningitis: Chronic meningitis with lymphocytic pleocytosis, elevated protein, low glucose on CSF
- Myelopathy: Transverse myelitis-like picture or progressive myelopathy
- Peripheral neuropathy: Axonal sensorimotor polyneuropathy, small fibre neuropathy (causing burning dysaesthesia)
- Pachymeningitis: Dural thickening mimicking meningioma
Investigations
- MRI brain with gadolinium — leptomeningeal enhancement, hypothalamic/periventricular T2 hyperintensity, cranial nerve enhancement
- Lumbar puncture — lymphocytic pleocytosis, elevated protein, ACE level in CSF (low sensitivity), low glucose
- FDG-PET/CT may identify occult systemic sarcoidosis to support the diagnosis
Management
Neurosarcoidosis generally warrants systemic immunosuppression due to the risk of irreversible neurological damage. High-dose oral prednisolone (0.5–1 mg/kg/day) is first-line, typically continued for several months before gradual tapering. Methotrexate or azathioprine serve as steroid-sparing agents. Infliximab has demonstrated efficacy in refractory neurosarcoidosis and should be considered early in aggressive presentations (e.g., myelopathy, hypothalamic involvement). Pregabalin or duloxetine may be adjunctive for small fibre neuropathy-related pain.
Granulomatous Inflammation & ACE
Sarcoidosis is defined by the presence of non-caseating granulomas — organised aggregates of activated macrophages (epithelioid cells), multinucleated giant cells, and surrounding T-lymphocytes. Understanding this pathological process and the role of angiotensin-converting enzyme (ACE) in the diagnostic work-up is essential for clinicians.
Granuloma Formation
The granulomatous response in sarcoidosis is driven by an exaggerated T-helper 1 (Th1) immune response. CD4+ T-lymphocytes release interleukin-2 (IL-2) and interferon-gamma (IFN-γ), recruiting and activating macrophages. These macrophages differentiate into epithelioid cells and fuse to form Langhans-type or foreign-body-type multinucleated giant cells. The granulomas may resolve completely or progress to fibrosis with hyalinisation.
- Immunological hallmark: CD4+ T-cell predominance in affected tissues, with a raised CD4/CD8 ratio in bronchoalveolar lavage (BAL) fluid (>3.5 is supportive)
- Non-caseating: Unlike tuberculosis, sarcoid granulomas lack central necrosis (caseation). However, fibrinoid necrosis may rarely be seen
- Schaumann bodies and asteroid bodies: Inclusions within giant cells — supportive but not pathognomonic
- Resolution vs. fibrosis: Approximately 60–70% of granulomas resolve; the remainder undergo fibrotic transformation, leading to organ dysfunction
Serum ACE — Role & Limitations
Angiotensin-converting enzyme is produced by epithelioid cells within sarcoid granulomas. Serum ACE (sACE) levels reflect the granuloma burden and disease activity.
| Feature | Detail |
|---|---|
| Sensitivity | ~60% (range 40–80%) — normal ACE does not exclude sarcoidosis |
| Specificity | ~90% in appropriate clinical context |
| False elevations | TB, leprosy, Gaucher disease, histoplasmosis, silicosis, berylliosis, hyperthyroidism, diabetes mellitus |
| ACE inhibitor effect | ACE inhibitors lower sACE — interpret with caution; consider temporarily withholding if measurement needed |
| ACE insertion/deletion polymorphism | DD genotype has higher baseline ACE; genotype-corrected reference ranges improve accuracy but are not yet standard in Australia |
| Monitoring role | Serial sACE may correlate with disease activity and treatment response, but is unreliable as a sole monitor |
Other Laboratory Findings
- Serum calcium: Hypercalcaemia (~10–13%) and hypercalciuria (~40%) — granulomas produce 1,25-dihydroxyvitamin D (calcitriol) independent of PTH regulation
- Raised ESR and CRP: Non-specific; elevated in active disease
- Lymphopenia: Common; may reflect sequestration of CD4+ cells in granulomas
- Raised alkaline phosphatase: Suggests hepatic granulomatous involvement
- Lysozyme: Raised; similar utility to ACE but not widely available in Australian laboratories
Clinical Presentation & Diagnostic Criteria
Typical Presentations by Organ System
| Organ | Frequency | Manifestations |
|---|---|---|
| Lungs | >90% | Cough, dyspnoea, bilateral hilar lymphadenopathy |
| Skin | 25–35% | Plaques, papules, lupus pernio, erythema nodosum |
| Eyes | 20–30% | Anterior uveitis, posterior uveitis, conjunctival nodules |
| Lymph nodes | 15–40% | Bilateral hilar, peripheral, mediastinal lymphadenopathy |
| Heart | 5% (clinically), 25% (autopsy) | Heart block, VT, cardiomyopathy, sudden death |
| Nervous system | 5–15% | Facial palsy, diabetes insipidus, myelopathy |
| Liver | 10–20% | Hepatomegaly, raised ALP, rarely cirrhosis |
| Joints | 10–15% | Ankle arthritis (Lofgren), chronic arthropathy |
Diagnostic Criteria
Diagnosis of sarcoidosis requires a consistent clinical and radiological picture supported by histological evidence of non-caseating granulomas, with exclusion of other causes of granulomatous inflammation. The following three criteria must be met:
- Compatible clinical and/or radiological features
- Histological demonstration of non-caseating granulomas on biopsy
- Exclusion of other granulomatous diseases — tuberculosis (AFB stain and culture, TB PCR), fungal infections (special stains, culture), berylliosis, foreign body reaction, malignancy, vasculitis
Biopsy Sites — When Is Tissue Required?
- Not always required: Lofgren syndrome with classical triad; Stage I BHL with typical presentation in a young adult
- Always required: Atypical presentations, Stage III/IV disease, suspected malignancy, treatment-refractory disease
- Preferred biopsy sites: Skin (easily accessible), peripheral lymph nodes, bronchoscopic mucosal biopsy or transbronchial lung biopsy (diagnostic yield ~80%), conjunctival nodules
- Endobronchial ultrasound (EBUS)-guided mediastinal lymph node sampling: Increasingly used in Australian centres; high diagnostic yield for mediastinal/hilar disease

Investigations
Baseline Work-Up for All Patients
Risk Stratification & Severity
Risk stratification in sarcoidosis guides the decision to observe, treat, or escalate to advanced therapies. The following severity framework incorporates organ involvement, disease trajectory, and prognostic factors.
Poor Prognostic Factors
- African American ethnicity (higher mortality, more aggressive disease)
- Stage III or IV radiographic disease
- Cardiac or neurological involvement
- Lupus pernio (associated with chronic disease course)
- Progressive pulmonary fibrosis (Stage IV)
- Hypercalcaemia persisting beyond initial presentation
- Sarcoidosis-associated pulmonary hypertension
Treatment — Steroids, Methotrexate & Infliximab
Treatment of sarcoidosis is indicated for patients with significant symptoms, progressive disease, organ-threatening involvement, or declining organ function. The decision to treat balances the potential benefits of immunosuppression against corticosteroid side effects and drug toxicity. Asymptomatic Stage I disease and self-limiting Lofgren syndrome typically do not require pharmacotherapy.
First-Line: Oral Corticosteroids
Steroid-Sparing Agents
Methotrexate is the preferred first-line steroid-sparing agent in Australian practice. It should be initiated early when prolonged corticosteroid therapy is anticipated (≥3–6 months), or as monotherapy in steroid-intolerant patients.
Biologic Therapy — Infliximab
Infliximab, a TNF-α monoclonal antibody, has robust evidence for refractory sarcoidosis, including pulmonary, neurosarcoidosis, cardiac, and cutaneous (lupus pernio) disease. It is generally reserved for patients who have failed or are intolerant of corticosteroids plus at least one conventional steroid-sparing agent.
Treatment Algorithm Summary
Monitoring
Monitoring in sarcoidosis encompasses disease activity assessment, organ function surveillance, and treatment toxicity screening. A structured follow-up plan enables early detection of relapse and drug-related adverse effects.
| Parameter | Frequency | Purpose |
|---|---|---|
| Chest X-ray | Every 3–6 months (first 2 years); then annually | Radiographic staging, progression detection |
| Spirometry + DLCO | Every 3–6 months during active disease; annually if stable | FVC trend — ≥10% decline triggers treatment initiation or escalation |
| Serum ACE | Every 3–6 months if elevated at baseline | Trend monitoring (supplementary to clinical/radiological assessment) |
| Serum calcium, creatinine | Every 3–6 months | Hypercalcaemia detection, renal function |
| FBC, LFTs | Every 1–3 months on DMARDs | Drug toxicity monitoring |
| ECG | Baseline and annually; more frequently if cardiac sarcoidosis suspected | Conduction abnormalities, arrhythmias |
| Ophthalmological review | Baseline and if symptoms develop (visual change, eye pain, red eye) | Anterior uveitis screening |
| DEXA scan | Baseline if prednisolone ≥3 months planned; repeat at 1–2 years | Osteoporosis risk from corticosteroids; consider bisphosphonate prophylaxis |
| CMR or PET-CT | As clinically indicated for cardiac or refractory disease | Active inflammation mapping, treatment response |
When to Re-Refer or Escalate
- FVC decline ≥10% despite treatment
- New cardiac symptoms (palpitations, syncope, heart failure signs)
- New neurological symptoms (facial weakness, visual loss, limb weakness)
- Refractory hypercalcaemia
- Drug toxicity or intolerance to DMARDs
Special Populations
Aboriginal and Torres Strait Islander Health
📚 References
- 1. Grunewald J, Grutters JC, Arkema EV, et al. Sarcoidosis. Nature Reviews Disease Primers. 2019;5(1):45.
- 2. Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis. European Respiratory Journal. 2021;58(6):2004079.
- document.querySelectorAll('.topic-toc a[href^="#"]').forEach(o=>{o.addEventListener("click",i=>{const t=o.getAttribute("href")||"",e=document.querySelector(t);if(!e)return;i.preventDefault();const n=e.getBoundingClientRect().top+window.scrollY-70;window.scrollTo({top:n,behavior:"smooth"}),history.replaceState(null,"",t)})});const r=new Map;document.querySelectorAll(".topic-toc a").forEach(o=>{const i=o.getAttribute("href")?.slice(1);i&&r.set(i,o)});if(r.size>0&&"IntersectionObserver"in window){const o=new IntersectionObserver(i=>{let t=null;for(const e of i)if(e.isIntersecting){t=e.target.id;break}t&&r.forEach((e,n)=>e.classList.toggle("active",n===t))},{rootMargin:"-80px 0px -40% 0px",threshold:0});r.forEach((i,t)=>{const e=document.getElementById(t);e&&o.observe(e)})}window.matchMedia("(max-width: 900px)").matches&&document.querySelector(".topic-rail-collapsible")?.removeAttribute("open");const c=document.querySelector(".topic-rail-details");function s(){c&&(window.matchMedia("(max-width: 900px)").matches?c.open=!1:c.open=!0)}s();let l;window.addEventListener("resize",()=>{clearTimeout(l),l=window.setTimeout(s,120)});