📋 Key Information Summary
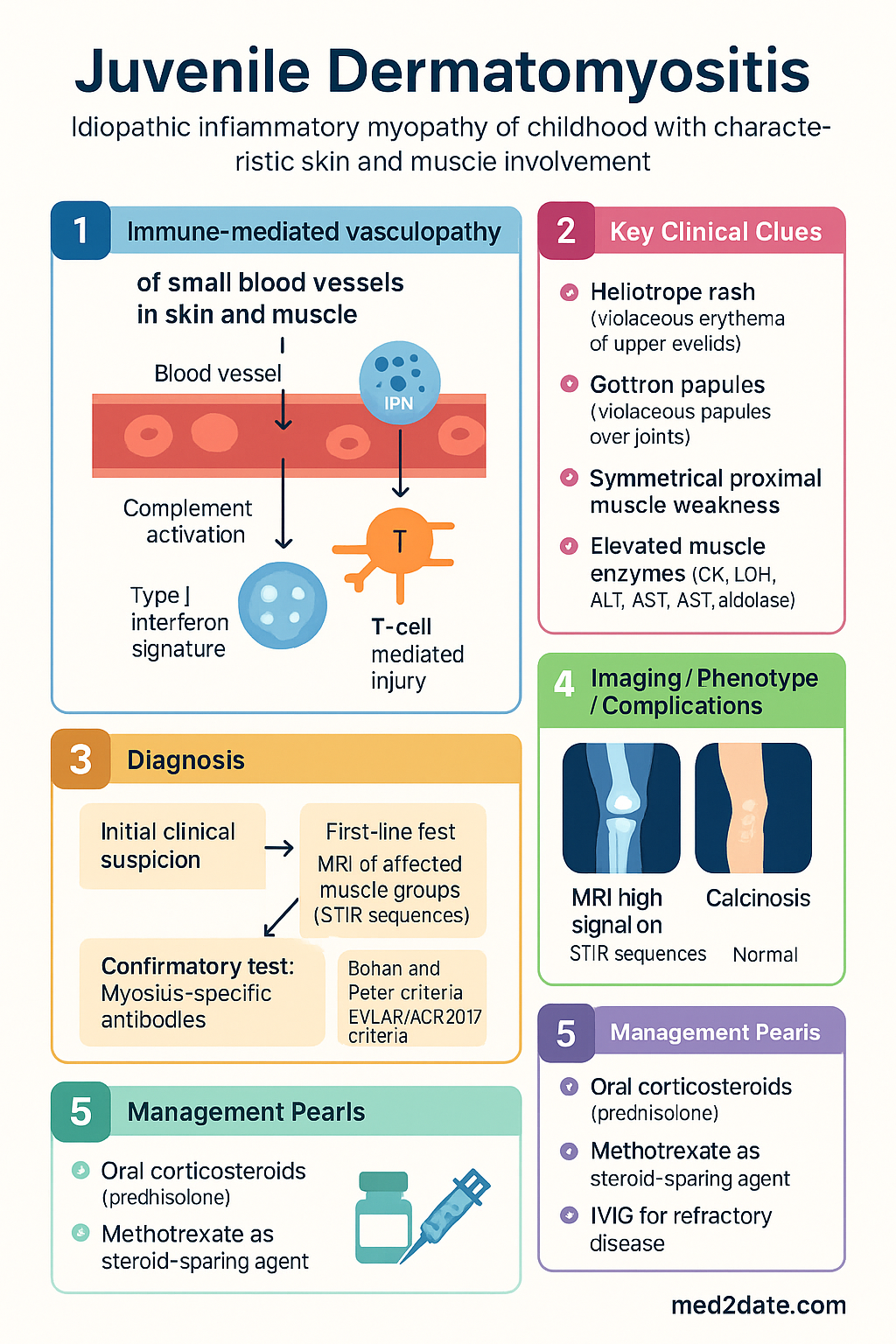
- Juvenile dermatomyositis (JDM) is the most common idiopathic inflammatory myopathy of childhood, characterised by immune-mediated vasculopathy of small blood vessels in skin and muscle; median onset age 7 years (range 2–15 years).
- Diagnostic triad: characteristic skin disease (heliotrope rash, Gottron papules), symmetrical proximal muscle weakness, and elevated muscle enzymes (CK, LDH, ALT, AST, aldolase).
- Heliotrope rash — violaceous erythema of the upper eyelids with possible periorbital oedema — is pathognomonic; Gottron papules — violaceous papules over MCP, PIP, DIP, elbow and knee extensor surfaces — are highly specific.
- Proximal muscle weakness typically develops insidiously over weeks to months; present as difficulty climbing stairs, rising from a seated position, or combing hair.
- Calcinosis occurs in 20–40% of Australian paediatric JDM patients and is associated with delayed or inadequate treatment; it may present as subcutaneous nodules, tumoural deposits, or dystrophic calcification.
- MRI of affected muscle groups (STIR sequences) is the preferred imaging modality to assess oedema, guide biopsy site, and monitor treatment response — available at all Australian tertiary paediatric centres.
- Myositis-specific antibodies (anti-Mi-2, anti-TIF1-γ, anti-NXP-2, anti-MDA5, anti-SAE) help classify phenotype, predict complications, and guide monitoring; anti-NXP-2 is most strongly associated with calcinosis.
- First-line treatment: oral corticosteroids (prednisolone 2 mg/kg/day, max 60 mg) with early introduction of methotrexate as steroid-sparing agent — methotrexate is PBS-listed (Restricted Benefit) for JDM.
- IVIG (Intragam® or Privigen®) is recommended as add-on therapy for refractory disease or significant swallowing difficulty; typically 2 g/kg divided over 2–5 days, repeated monthly.
- Biologic agents (rituximab, tocilizumab) are reserved for refractory JDM unresponsive to conventional therapy; rituximab is available via PBS Authority for severe autoimmune disease.
- Monitor for calcinosis, lipodystrophy, growth impairment, and steroid-related complications (osteoporosis, avascular necrosis, adrenal insufficiency, glucose intolerance).
- Aboriginal and Torres Strait Islander children may experience delayed diagnosis due to geographic barriers to specialist care; targeted education and telehealth rheumatology are essential.
Introduction & Australian Epidemiology
Juvenile dermatomyositis (JDM) is a rare, chronic autoimmune inflammatory myopathy of childhood and adolescence, defined by characteristic cutaneous manifestations and symmetrical proximal muscle inflammation. It accounts for approximately 85% of all idiopathic inflammatory myopathies in children, with juvenile polymyositis representing the remainder.
JDM is classified using the Bohan and Peter criteria (1975), subsequently revised by the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) 2017 criteria, which incorporate myositis-specific antibodies and MRI findings. The pathogenesis involves immune-mediated vasculopathy of small blood vessels (capillary, arteriole, venule) in skin and skeletal muscle, driven by complement activation (membrane attack complex deposition), type I interferon signature, and T-cell mediated injury.
In Australia, the estimated incidence is 2–4 per million children per year, with approximately 20–40 new cases diagnosed nationally each year. The Australian Paediatric Rheumatology Group (APRG) registry data suggest a female-to-male ratio of approximately 2–3:1, with peak onset between 5 and 10 years of age. There is no significant difference in incidence between metropolitan and regional populations, although Aboriginal and Torres Strait Islander children in remote communities may face diagnostic delays due to limited access to specialist rheumatology services.
The prognosis of JDM has improved substantially over the past three decades with earlier recognition, aggressive treatment protocols, and better supportive care. However, significant morbidity persists: calcinosis affects 20–40% of patients, lipodystrophy 10–25%, and chronic disease course with flares occurs in approximately 30–50% of cases. Mortality is now below 2% in developed nations, primarily from gastrointestinal vasculopathy or interstitial lung disease.
Heliotrope & Gottron Rash
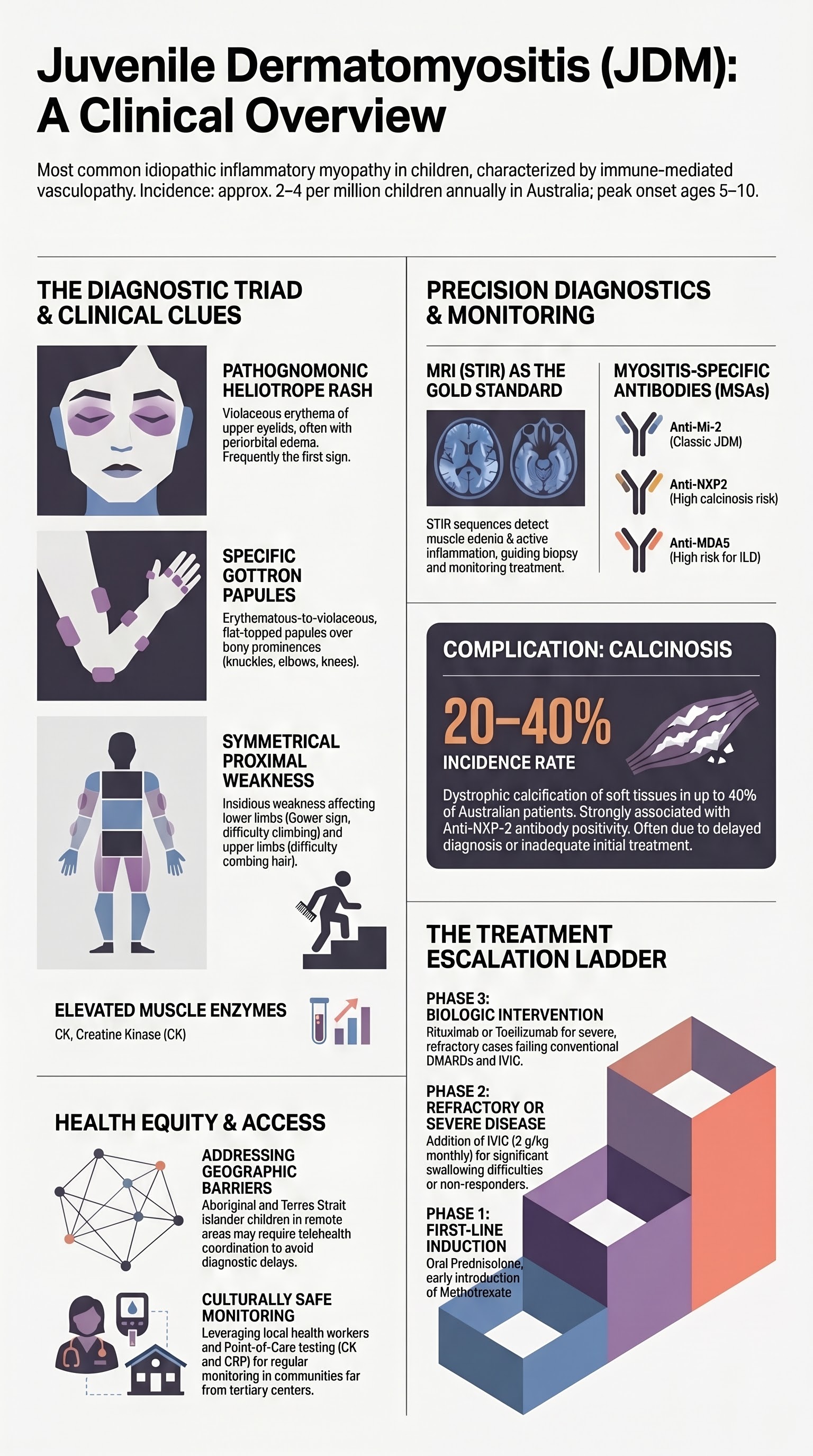
Cutaneous manifestations are frequently the initial presenting feature of JDM and may precede muscle weakness by weeks to months. The heliotrope rash and Gottron papules are the hallmark dermatological findings, present in approximately 70–80% and 60–80% of JDM patients respectively.
Heliotrope Rash
The heliotrope rash is a violaceous (lilac-coloured) erythema affecting the upper eyelids, often with associated periorbital oedema. The colour is described as pathognomonic and is named after the heliotrope flower. It may extend to the malar area, forehead, and V-neck region. Telangiectasia of the nail folds is a related finding seen in up to 70% of patients.
- Distribution: Bilateral, upper eyelids (may extend to periorbital and malar skin)
- Colour: Violaceous, lilac, or dusky pink-purple erythema
- Associated features: Periorbital oedema (often the first sign noted by parents), telangiectasia of nail fold capillaries
- Differential diagnoses: Allergic contact dermatitis, atopic dermatitis, systemic lupus erythematosus (malar rash spares the nasolabial folds), erythema infectiosum
Gottron Papules
Gottron papules are erythematous-to-violaceous, flat-topped papules and plaques located over the bony prominences and extensor surfaces of the metacarpophalangeal (MCP) joints, proximal interphalangeal (PIP) joints, and distal interphalangeal (DIP) joints. They are pathognomonic for dermatomyositis when present and are not seen in other childhood rashes.
- Distribution: MCP, PIP, DIP joints; elbows; knees; medial malleoli
- Morphology: Flat-topped, erythematous-to-violaceous papules, may be scaly; may coalesce into plaques
- Gottron sign: Similar erythematous macular patches over extensor surfaces of large joints (elbows, knees) — less specific than Gottron papules
- Histopathology: Interface dermatitis with vacuolar degeneration of the basal layer, mucin deposition, and perivascular lymphocytic infiltrate
Other Cutaneous Features
Shawl sign (erythema over the upper back, shoulders, and posterior neck), V-sign (erythema of the anterior chest in a V distribution), mechanic's hands (hyperkeratosis and fissuring of the lateral fingers and palms), poikiloderma (telangiectasia, dyspigmentation, atrophy), and periungual nailfold capillary changes (dilated, bushy capillary loops with dropout areas) are additional characteristic findings.
Proximal Muscle Weakness
Symmetrical proximal muscle weakness is a core feature of JDM, present in 60–80% of patients at diagnosis. It develops insidiously over weeks to months and reflects inflammatory myopathy of the skeletal muscle.
Clinical Features
- Lower limb predominance: Difficulty climbing stairs, rising from the floor (Gower sign), getting out of a car, or stepping onto a kerb
- Upper limb involvement: Difficulty lifting arms above the head (combing hair, reaching overhead shelves), weakness with gripping
- Neck flexor weakness: Difficulty holding the head upright; child may lie with head supported; a useful bedside sign
- Pharyngeal and oesophageal weakness: Dysphagia, nasal regurgitation, dysphonia (hoarse voice); may be severe and is a poor prognostic sign
- Gait disturbance: Waddling gait, Trendelenburg sign, toe-walking due to calf contracture (late finding)
Functional Assessment
Standardised functional assessment is essential for disease monitoring. The Childhood Myositis Assessment Scale (CMAS) and Manual Muscle Testing (MMT-8) are validated tools used in Australian paediatric rheumatology centres:
| Assessment Tool | Components | Score Range | Interpretation |
|---|---|---|---|
| CMAS | 21 items assessing function, strength, endurance (floor rise, arm raise, sit-ups, step-ups, etc.) | 0–52 | ≤15 = severe weakness; 15–40 = moderate; >40 = mild/remission |
| MMT-8 | Manual muscle testing of 8 muscle groups (neck flexors, deltoid, biceps, wrist extensors, gluteus maximus, gluteus medius, quadriceps, ankle dorsiflexors) | 0–80 | Higher = better strength; serial measurement to track response |
Calcinosis
Calcinosis (dystrophic calcification of soft tissues) is a significant long-term complication of JDM, occurring in 20–40% of Australian paediatric patients. It is a major source of morbidity, causing pain, functional limitation, skin ulceration, secondary infection, and joint contractures. Calcinosis typically develops 1–3 years after disease onset but may appear earlier in severe, refractory, or undertreated disease.
Risk Factors
- Delayed diagnosis and treatment initiation (>4 months from symptom onset)
- Persistent or relapsing disease course with ongoing muscle enzyme elevation
- Anti-NXP-2 antibody positivity (strongest serological association with calcinosis)
- Severe skin disease (extensive ulceration, widespread Gottron papules)
- Younger age at onset (<5 years in some series)
- Gastrointestinal vasculopathy at presentation
Clinical Patterns
| Pattern | Description | Common Sites |
|---|---|---|
| Superficial nodular | Small, firm, subcutaneous nodules; may ulcerate through skin with chalky discharge | Elbows, knees, fingers, over pressure points |
| Deep nodular / tumoural | Large, deep deposits in muscles and fascia; painful, may restrict movement | Proximal limbs, trunk, buttocks |
| Plaque-like | Sheet-like calcific deposits under the skin | Trunk, thighs |
| Exoskeleton / generalised | Extensive, armour-like calcification encasing limbs or trunk; most debilitating | Widespread |
Management of Calcinosis
No single therapy has robust evidence for calcinosis regression; treatment is based on case series and expert consensus:
- Ensure disease remission: Optimal immunosuppression to achieve and maintain remission is the single most important preventive measure
- Diltiazem: 3–5 mg/kg/day PO in divided doses; may slow progression and promote regression — used off-label in paediatrics
- Colchicine: 0.02–0.03 mg/kg/day PO (max 1 mg/day); anti-inflammatory effect may help — limited evidence
- IVIG: May have a role in refractory calcinosis when combined with immunosuppression
- Sodium thiosulphate: IV or topical; emerging evidence for chelation of calcium deposits
- Surgical excision: Reserved for symptomatic, localised deposits causing functional impairment or recurrent infection
- Low-dose warfarin: 0.05–0.1 mg/kg/day; reported to reduce progression in some series — monitor INR closely
MRI Muscle Imaging
MRI of the affected muscle groups is the gold-standard non-invasive imaging modality for assessing inflammatory myopathy in JDM. It is available at all Australian tertiary paediatric centres and most major metropolitan hospitals. MRI is used for diagnosis (demonstrating active inflammation), guiding muscle biopsy site selection, and serial monitoring of treatment response.
MRI Sequences and Findings
| Sequence | Key Finding in Active JDM | Clinical Utility |
|---|---|---|
| STIR (Short Tau Inversion Recovery) | High signal (bright) in inflamed muscles — represents oedema from active myositis | Most sensitive for active disease; primary monitoring sequence |
| T1-weighted | Fatty infiltration (high signal) in chronic disease; muscle atrophy | Assesses chronic damage; poor prognostic sign if extensive |
| T1 with gadolinium contrast | Enhancement of inflamed muscles and fascial planes | Confirms active inflammation; guides biopsy site |
| T2-weighted | Increased signal in oedematous muscles | Complementary to STIR; less sensitive for subcutaneous oedema |
Muscle Groups to Image
- Lower limbs: Quadriceps, hamstrings, adductors, gluteal muscles, calves — most commonly affected
- Upper limbs: Deltoids, biceps, triceps, forearm extensors — if clinically indicated
- Pelvis and thighs: Preferred initial site as it captures the largest muscle groups and is most sensitive for early disease
- Whole-body MRI: Increasingly used in specialised centres; captures perifascicular oedema pattern characteristic of dermatomyositis
MBS Item Numbers
MRI of the affected limb(s) is billed under MBS item numbers for MRI of the relevant body region. Paediatric sedation or general anaesthesia services are billed separately. Bulk-billing arrangements are typically available at public tertiary paediatric hospitals.
Myositis-Specific Antibodies in Children
Myositis-specific antibodies (MSAs) are detectable in approximately 60–80% of JDM patients and are increasingly used to classify disease phenotype, predict organ-specific complications, and guide individualised management. MSA testing is available through major Australian immunopathology laboratories (e.g., Royal Prince Alfred Hospital, The Royal Children's Hospital Melbourne) and reference laboratories.
| Antibody | Frequency in JDM | Clinical Phenotype | Key Associations |
|---|---|---|---|
| Anti-Mi-2 | 10–30% | Classic JDM: prominent skin disease with classic heliotrope and Gottron rash; good muscle response to treatment | Better prognosis; lower risk of calcinosis; responsive to standard therapy |
| Anti-TIF1-γ (p155/140) | 20–35% | Prominent skin disease; malignancy-associated in adults (no malignancy association in children) | Severe skin ulceration; photosensitivity; lipodystrophy; moderate calcinosis risk |
| Anti-NXP2 (MJ/p140) | 15–25% | Calcinosis-predominant phenotype; may have more muscle disease | Strongest association with calcinosis; gastrointestinal vasculopathy; younger age at onset |
| Anti-MDA5 (CADM-140) | 5–15% | Clinically amyopathic (minimal muscle weakness); severe skin ulceration; arthralgia | Interstitial lung disease (ILD) — screen with HRCT and PFTs; oral ulceration; skin ulceration |
| Anti-SAE | 2–5% | Dysphagia; skin disease preceding muscle involvement | Dysphagia; less common in children than adults |
| Anti-Jo-1 (and other anti-synthetase) | 2–5% | Antisynthetase syndrome: ILD, arthritis, Raynaud, mechanic's hands, fever | Interstitial lung disease; rare in pure JDM; more polymyositis overlap |
Clinical Utility of MSA Testing
- Diagnostic confirmation: Supports JDM diagnosis, particularly in seronegative or clinically amyopathic presentations
- Risk stratification: Anti-NXP2 → monitor closely for calcinosis; anti-MDA5 → screen for ILD with HRCT at diagnosis
- Treatment decisions: Anti-MDA5-positive patients with ILD may require more aggressive therapy (rituximab, cyclophosphamide)
- Prognostic information: Anti-Mi-2 generally associated with better outcomes; anti-NXP2 with higher calcinosis risk
- Seronegative JDM: ~20–40% of patients are MSA-negative; diagnosis is based on clinical, CK, MRI, and biopsy criteria
Treatment: Steroids, MTX, IVIG, and Biologics
The treatment of JDM aims to achieve rapid disease control, prevent organ damage (particularly calcinosis and contractures), and minimise corticosteroid toxicity. A stepwise, treat-to-target approach is recommended by the Australian Paediatric Rheumatology Group and the Single Hub and Access Point for Paediatric Rheumatology in Europe (SHARE) initiative.
First-Line Therapy: Corticosteroids
Steroid-Sparing Agent: Methotrexate
Second-Line / Add-On: Intravenous Immunoglobulin (IVIG)
Biologic Agents (Refractory Disease)
Treatment Algorithm
Adjunctive Measures
- Calcium and vitamin D: All patients on corticosteroids — calcium supplementation (age-appropriate dose) + cholecalciferol 400–800 IU/day PO
- Bisphosphonates: Consider for osteoporosis prevention if prolonged steroid use; discuss with paediatric endocrinologist
- Pneumocystis jirovecii prophylaxis: Co-trimoxazole (trimethoprim 2–5 mg/kg/day PO, 3 days/week) if on high-dose prednisolone + MTX combination; or at least while on prednisolone >0.5 mg/kg/day
- Sun protection: Strict — broad-brimmed hat, SPF 50+ sunscreen, UPF 50+ clothing; photosensitivity is common in JDM and exacerbates skin disease
- Physiotherapy: Active range-of-motion exercises from disease onset; progressive strengthening once inflammation controlled; avoid overexertion during active disease
- Psychosocial support: Referral to clinical psychologist; school support; young person support groups (e.g., Arthritis Australia)
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Enders FB, Bader-Meunster B, Sanner H, et al. Recommendations for the management of juvenile dermatomyositis — an international, multicentre SHARE initiative. Rheumatology (Oxford). 2024;63(3):673–689.
- 2. Rider LG, Ruperto N, Pistorio A, et al. 2016 ACR/EULAR provisional criteria for minimal disease activity in the adult and juvenile idiopathic inflammatory myopathies and their major organ damage. Ann Rheum Dis. 2017;76(12):1987–1995.
- 3. Papadopoulou C, McCann LJ. The risk of calcification in juvenile dermatomyositis: a review. Curr Rheumatol Rep. 2021;23(10):76.
- 4. Tansley SL, Betteridge ZE, Shaddick G, et al. Anti-MDA5 juvenile dermatomyositis is associated with a more severe phenotype and higher rates of calcinosis. Ann Rheum Dis. 2014;73(4):e27.
- 5. Ruperto N, Pistorio A, Oliveira S, et al. Prednisone versus prednisone plus cyclosporine versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet. 2016;387(10019):671–678.
- 6. Lam CG, Manlhiot C, Pullenayegum EM, Feldman BM. Efficacy of intravenous immunoglobulin therapy in juvenile dermatomyositis. Ann Rheum Dis. 2011;70(3):437–442.
- 7. Agarwal S, Dugan JL, De Paolis C, et al. Use of rituximab in refractory juvenile dermatomyositis. Rheumatology (Oxford). 2020;59(8):1944–1951.
- 8. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2023. Canberra: AIHW; 2023.
- 9. McCann LJ, Pilkington CA, Huber AM, et al. Development of a consensus core dataset in juvenile dermatomyositis for clinical use to inform research. Ann Rheum Dis. 2018;77(7):990–997.
- 10. Mamyrova G, Rider LG, Haagenson L, et al. Myositis-specific antibodies in juvenile dermatomyositis: associations with clinical features and outcomes. Arthritis Rheumatol. 2022;74(10):1703–1713.
- 11. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–347.
- 12. Huber AM, Giannini EH, Lovell DJ, et al. Development of validated disease activity and damage measures for juvenile dermatomyositis. Arthritis Care Res. 2010;62(9):1240–1246.
- 13. Royal Australasian College of Physicians (RACP). Position statement on the use of telehealth in paediatric care. Sydney: RACP; 2022.
- 14. Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 2017;69(12):2271–2282.