📋 Key Information Summary
- IgA Vasculitis (IgAV, Henoch-Schönlein Purpura) is the most common systemic small-vessel vasculitis in children, predominantly affecting those aged 3–10 years.
- Pathophysiology involves IgA1-dominant immune complex deposition in vessel walls, triggered most often by upper respiratory tract infection (especially Group A Streptococcus).
- Clinical tetrad: (1) palpable purpura — mainly buttocks and lower extremities, (2) arthritis/arthralgia — large joints (knees, ankles), (3) colicky abdominal pain with GI bleeding risk, (4) renal involvement — haematuria, proteinuria, nephritis.
- Diagnosis is clinical using the EULAR/PReS 2010 classification criteria: palpable purpura (mandatory) plus at least one of diffuse abdominal pain, IgA deposition on biopsy, arthritis/arthralgia, or renal involvement.
- Skin biopsy showing leucocytoclastic vasculitis with IgA deposits on immunofluorescence confirms the diagnosis; renal biopsy is reserved for persistent nephritis or nephrotic-range proteinuria.
- The majority of children are managed supportively — disease is self-limiting in 95% of paediatric cases within 4–6 weeks.
- NSAIDs (ibuprofen) provide analgesia for arthralgia; avoid in active GI bleeding or renal impairment.
- Prednisolone 1 mg/kg/day (max 40 mg) for 1–2 weeks then taper is indicated for severe abdominal pain, intussusception risk, or significant renal involvement.
- For crescentic IgA nephritis (≥50% crescents on biopsy): IV methylprednisolone pulses, mycophenolate or cyclophosphamide, and consider plasmapheresis.
- Adult-onset IgAV carries a significantly worse renal prognosis — up to 30–40% develop chronic kidney disease versus 1–5% in children.
- Aboriginal and Torres Strait Islander children have higher rates of post-streptococcal glomerulonephritis overlap; early renal screening and culturally safe follow-up are essential.
- Long-term renal monitoring: urinalysis and BP at 1, 3, 6, 12 months then annually for at least 5 years; nephrology referral if persistent haematuria/proteinuria.
- Intussusception (usually ileo-ileal, not ileo-caecal) is the most serious GI complication; ultrasound is the preferred imaging modality.
- All patients should have throat culture/ASOT and renal function tests at diagnosis; MBS item 69309 (urinalysis) and 65070 (eGFR) are relevant for follow-up.
Introduction & Australian Epidemiology
IgA Vasculitis (IgAV), historically known as Henoch-Schönlein Purpura (HSP), is the most common systemic vasculitis of childhood and the most frequent cause of non-thrombocytopenic palpable purpura in the paediatric population. The condition is characterised by IgA1-dominant immune complex deposition within small vessel walls, leading to a triad of cutaneous, musculoskeletal, gastrointestinal, and renal manifestations.
In Australia, IgAV has an annual incidence of approximately 15–20 per 100,000 children under 16 years, with a peak incidence in autumn and winter months correlating with seasonal upper respiratory tract infections (URTIs). The disease is most common in children aged 3–10 years, with a male-to-female ratio of approximately 1.5:1. While paediatric cases dominate the epidemiological picture, adult-onset IgAV, though rarer (incidence ~1–2 per 100,000 adults), presents a more aggressive course, particularly regarding renal outcomes.
Approximately 50–75% of paediatric cases are preceded by an identifiable trigger — most commonly a Group A Streptococcal (GAS) pharyngitis or other URTI — within 1–3 weeks before symptom onset. Other documented triggers include medications (ACE inhibitors, certain antibiotics), vaccinations (influenza, hepatitis B), insect bites, and less commonly, malignancy in adults. The disease is more prevalent in Asian, Mediterranean, and Middle Eastern populations.
In the Australian context, particular attention must be paid to Aboriginal and Torres Strait Islander children, who experience higher rates of GAS infection and post-streptococcal complications in remote and regional communities. Close liaison with RHDAustralia guidelines and the Royal Darwin Hospital renal unit protocols is recommended for these populations.
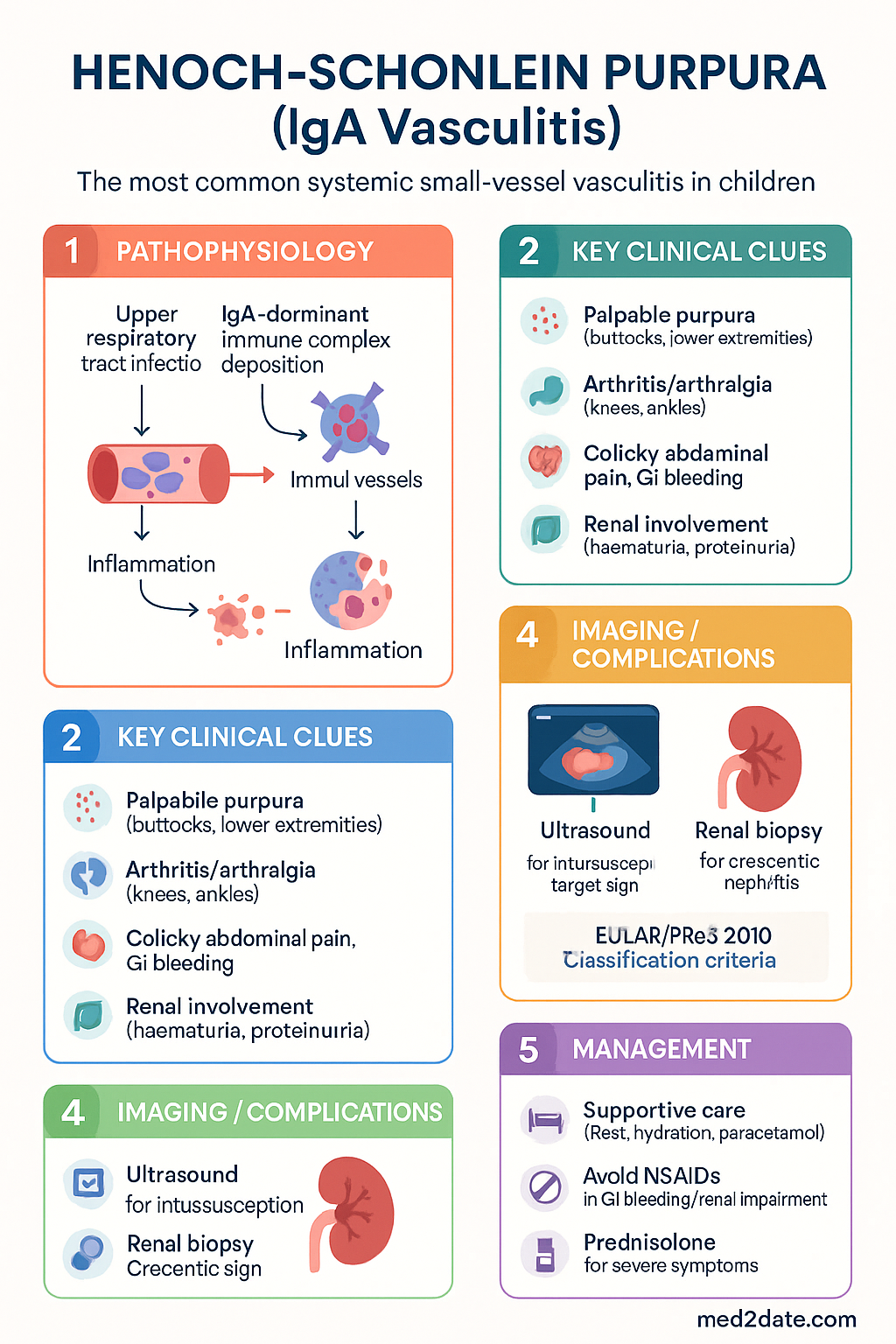
Pathophysiology
IgA Vasculitis is a leucocytoclastic vasculitis mediated by IgA1-containing immune complexes. The pathogenesis involves three interrelated mechanisms:
- Abnormal IgA1 glycosylation: Patients produce IgA1 with aberrantly galactosylated hinge-region O-linked glycans (galactose-deficient IgA1, or Gd-IgA1). This mirrors the defect seen in IgA nephropathy (Berger disease), suggesting a shared underlying immunological predisposition.
- Immune complex formation: Gd-IgA1 complexes with anti-glycan IgG autoantibodies, forming pathogenic immune complexes that deposit in small vessel walls — particularly in dermal capillaries, mesenteric arterioles, and the glomerular mesangium.
- Complement activation and inflammation: Deposited immune complexes activate the alternative complement pathway (and possibly lectin pathway), recruiting neutrophils and causing leucocytoclastic vasculitis with fibrinoid necrosis of arteriolar walls.
The inciting stimulus — commonly a respiratory tract pathogen — is believed to trigger an exaggerated mucosal IgA immune response. GAS pharyngitis is the most frequently implicated trigger in Australian practice, though Mycoplasma pneumoniae, parvovirus B19, adenovirus, and Haemophilus influenzae have also been documented. The seasonal clustering of IgAV in autumn/winter aligns with peak GAS and viral URTI transmission periods in temperate Australian climates.
Clinical Presentation & Diagnostic Criteria
Classic Clinical Tetrad
IgAV classically presents with a tetrad of clinical features, though not all are invariably present at initial assessment:
| Manifestation | Frequency | Key Features |
|---|---|---|
| Palpable purpura | 100% (mandatory for diagnosis) | Symmetric, dependent distribution — buttocks, lower extremities, ankles. May extend to upper limbs and trunk. Non-blanching, raised, may coalesce. No thrombocytopenia. |
| Arthritis / Arthralgia | 60–80% | Migratory, non-deforming. Large joints — knees, ankles most common. Self-limiting, no permanent damage. |
| Gastrointestinal | 50–75% | Colicky periumbilical/epigastric pain. Nausea, vomiting, GI bleeding (melaena, haematochezia). Intussusception risk (usually ileo-ileal). |
| Renal involvement | 20–60% | Haematuria (micro- or macroscopic), proteinuria, nephrotic syndrome, nephritic syndrome. Crescentic GN in severe cases. |
EULAR/PReS 2010 Classification Criteria
The European League Against Rheumatism (EULAR) / Paediatric Rheumatology European Society (PReS) criteria require:
- Diffuse abdominal pain
- Any biopsy showing predominant IgA deposition (skin or renal)
- Arthritis or arthralgia
- Renal involvement (haematuria and/or proteinuria)
Other Manifestations
- Scrotal oedema / orchitis: 2–35% of boys; usually painless, self-limiting.
- CNS involvement: Rare — headache, seizures, cerebral vasculitis.
- Pulmonary haemorrhage: Rare but life-threatening; presents with haemoptysis and acute respiratory failure.
- Testicular torsion mimicker: Important to differentiate from torsion urgently.
Investigations
IgAV is primarily a clinical diagnosis. Laboratory and histopathological investigations support the diagnosis, assess severity, and guide monitoring.
Baseline Investigations
Histopathology
- Skin biopsy (gold standard): Leucocytoclastic vasculitis with perivascular neutrophilic infiltrate and IgA deposits on direct immunofluorescence (DIF). Preferred site: early purpuric lesion on lower extremity. MBS 13020.
- Renal biopsy: Reserved for persistent nephrotic-range proteinuria, nephritic syndrome, declining eGFR, or suspected crescentic GN. Shows mesangial IgA deposits (IgA > C3 > IgG). Oxford classification (MEST-C) applies.