📋 Key Information Summary
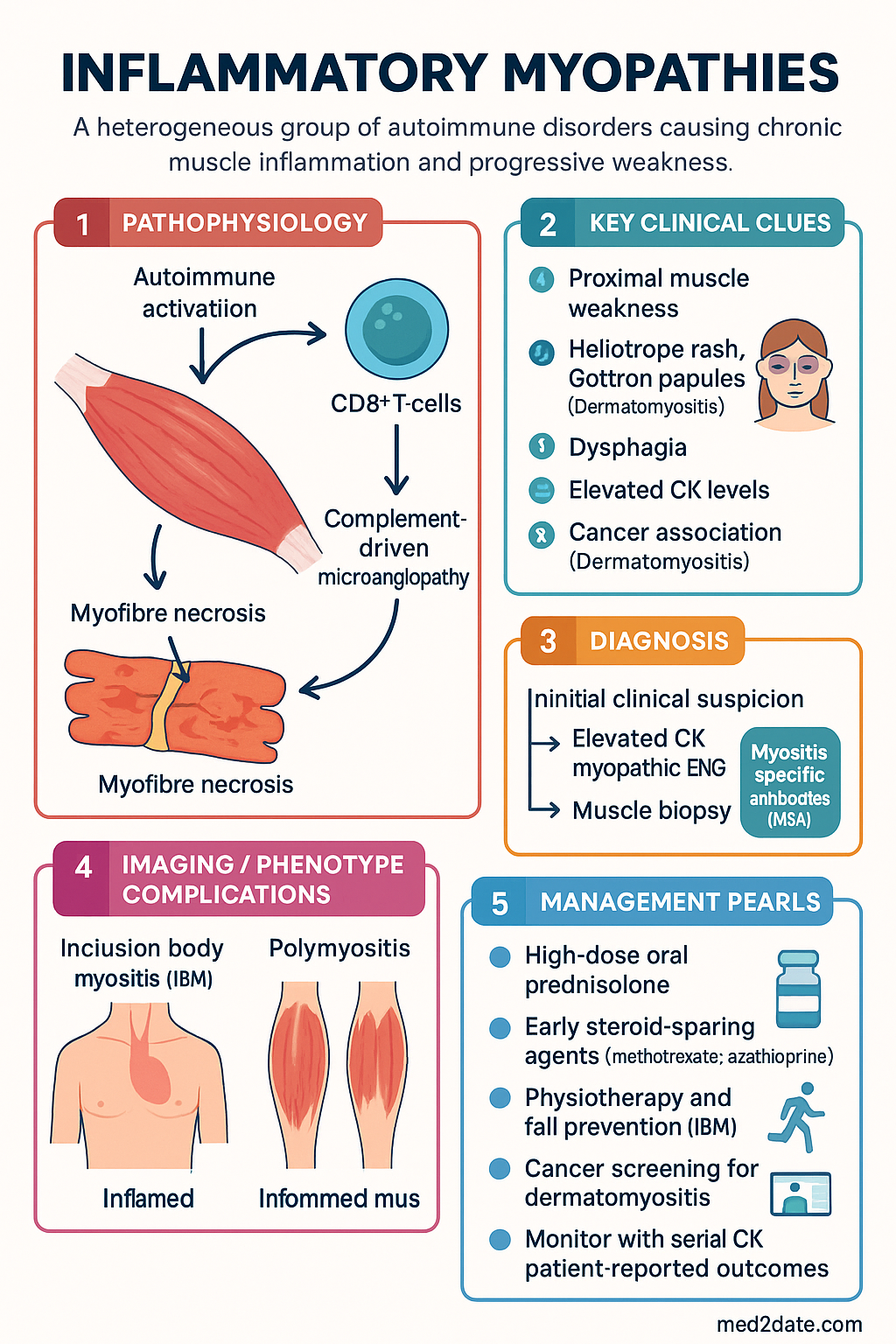
- Inflammatory myopathies are a heterogeneous group of autoimmune disorders causing chronic muscle inflammation and progressive weakness; major subtypes include dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM), and immune-mediated necrotising myopathy (IMNM).
- Dermatomyositis is characterised by proximal muscle weakness plus distinctive cutaneous features (heliotrope rash, Gottron papules) and carries a significant cancer association in adults — mandatory cancer screening within 3 years of diagnosis.
- Polymyositis presents with subacute proximal weakness without skin involvement; diagnosis requires elevated CK, myopathic EMG, and muscle biopsy showing endomysial CD8+ T-cell infiltrate.
- Inclusion body myositis is the most common acquired myopathy in patients over 50 years and is largely treatment-resistant; it characteristically affects finger flexors and quadriceps with dysphagia in up to 60%.
- Immune-mediated necrotising myopathy is strongly associated with anti-HMGCR (statin-exposed) and anti-SRP antibodies; features severe weakness, very high CK (often >10× ULN), and necrotising biopsy without significant inflammation.
- Myositis-specific antibodies (MSA) — including anti-Mi-2, anti-MDA5, anti-TIF1-γ, anti-NXP2, anti-SAE, anti-SRP, anti-HMGCR, and anti-synthetase antibodies — define distinct clinical phenotypes and cancer risk.
- First-line therapy for DM, PM, and IMNM is high-dose oral prednisolone 1 mg/kg/day (max 60 mg) with early steroid-sparing agent (methotrexate, azathioprine, or mycophenolate).
- IBM is largely refractory to conventional immunosuppression; physiotherapy, fall prevention, and dysphagia management are cornerstones; IVIg may provide modest benefit in selected patients.
- All adult-onset DM patients require age-appropriate malignancy screening (CT chest/abdomen/pelvis, pelvic exam, mammography, colonoscopy as indicated) within the first 3 months and continued surveillance for 3–5 years.
- Anti-MDA5 dermatomyositis carries high risk of rapidly progressive interstitial lung disease; early pulmonary function testing and HRCT are essential.
- Aboriginal and Torres Strait Islander patients may present later due to barriers to specialist access; culturally safe care pathways and remote telehealth rheumatology support are important.
- Monitor disease activity with serial CK, aldolase, muscle strength testing, and patient-reported outcomes; adjust steroid-sparing therapy to achieve steroid-free or low-dose steroid remission.
- Key drug interactions: methotrexate with trimethoprim (bone marrow suppression), azathioprine with allopurinol (myelosuppression); check TPMT before azathioprine.
Introduction & Australian Epidemiology
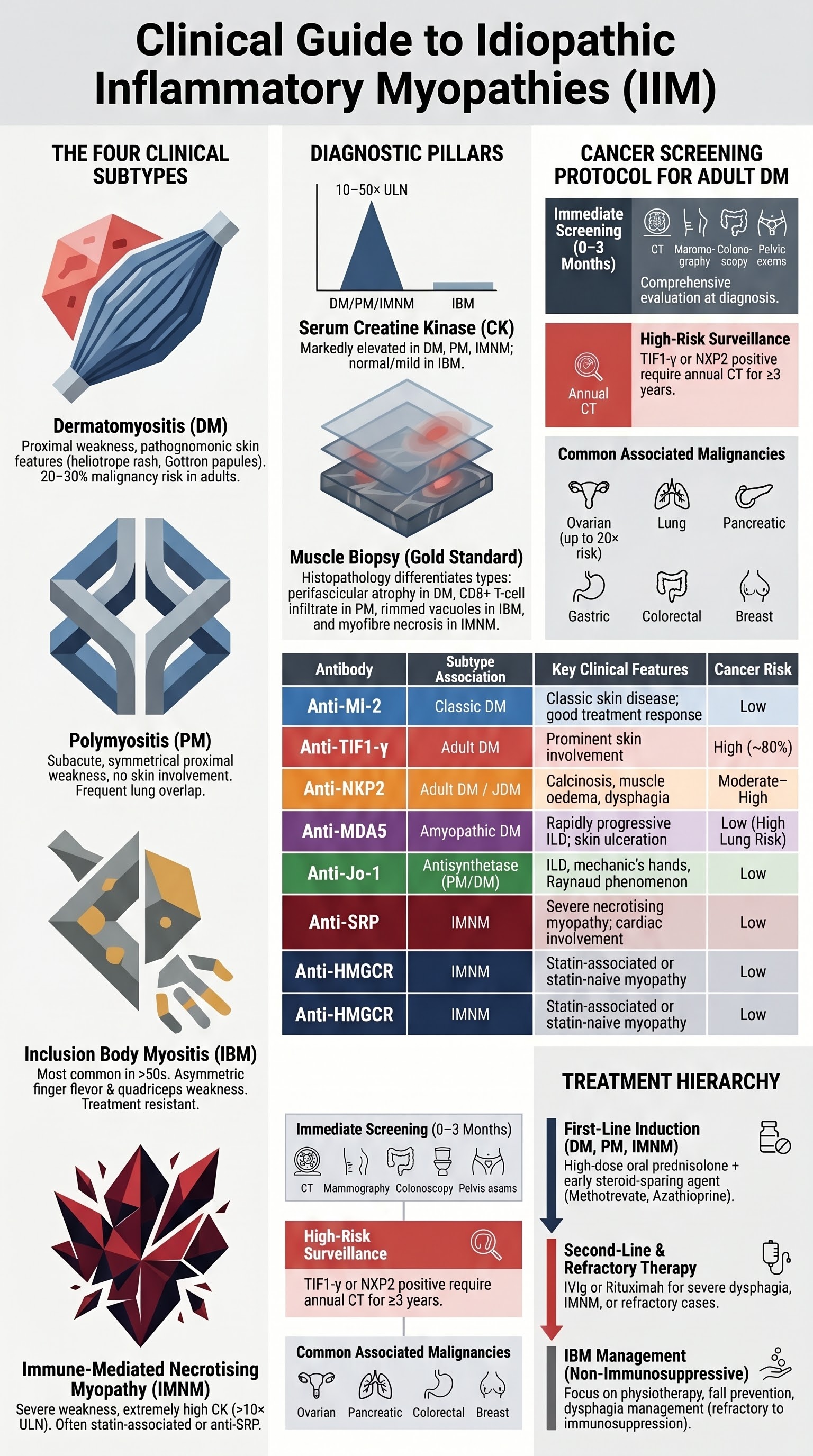
The idiopathic inflammatory myopathies (IIM) are a group of acquired autoimmune diseases characterised by chronic muscle inflammation, progressive skeletal muscle weakness, and multi-organ involvement. The principal adult subtypes — dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM), and immune-mediated necrotising myopathy (IMNM) — differ in pathogenesis, clinical phenotype, cancer association, and treatment responsiveness.
In Australia, population-based data from the Australian Institute of Health and Welfare (AIHW) and state-based registries estimate the overall prevalence of IIM at approximately 10–15 per 100,000 persons. Dermatomyositis has an annual incidence of approximately 5–9 per million, with a bimodal age distribution peaking in childhood (5–15 years) and adulthood (45–60 years). PM is rarer and increasingly recognised to overlap with other autoimmune conditions (overlap myositis). IBM is the most common acquired myopathy in patients over 50 years, with an estimated prevalence of 45–70 per million in those aged ≥50, and a male-to-female ratio of approximately 3:1.
IMNM has gained increasing recognition since the association of anti-HMGCR antibodies with statin exposure was described in 2010. With Australia's high rates of statin use (approximately 2.5 million PBS prescriptions annually), statin-associated IMNM is an important consideration in any patient presenting with unexplained myalgia and markedly elevated creatine kinase (CK).
The overlap of DM and PM with systemic autoimmune features — interstitial lung disease (ILD), arthritis, Raynaud phenomenon, and mechanic's hands — is collectively termed antisynthetase syndrome when associated with anti-aminoacyl-tRNA synthetase antibodies. Australian rheumatology referral pathways emphasise early recognition and MSA testing to guide cancer screening urgency and therapeutic decisions.
Dermatomyositis
Dermatomyositis is an autoimmune inflammatory myopathy with distinctive cutaneous manifestations and a well-established association with malignancy in adults. It is mediated by complement-driven microangiopathy affecting intramuscular blood vessels and skin.
Clinical Features
- Muscle: Symmetrical, progressive proximal weakness (difficulty rising from chairs, climbing stairs, lifting arms above head). Dysphagia occurs in 20–30%.
- Skin (pathognomonic): Heliotrope rash (violaceous periorbital oedema), Gottron papules (erythematous papules over MCP/PIP/DIP joints), shawl sign, V-sign, mechanic's hands, periungual erythema, and calcinosis (more common in juvenile DM).
- Systemic: Interstitial lung disease (especially anti-MDA5 and anti-synthetase), dysphagia, cardiac involvement (arrhythmia, myocarditis), arthralgia, constitutional symptoms.
Diagnosis
Diagnosis follows the 2017 EULAR/ACR classification criteria. Characteristic skin findings plus proximal weakness, elevated CK, and myopathic EMG or MRI signal abnormality strongly suggest DM. Muscle biopsy shows perifascicular atrophy — the histopathological hallmark.
Treatment
Cutaneous Management
- Sun protection (SPF 50+, protective clothing) — essential; UV exposure exacerbates skin disease.
- Topical corticosteroids (moderate potency, e.g., mometasone furoate 0.1%) for localised Gottron papules and rash.
- Hydroxychloroquine 200–400 mg/day PO — PBS General Benefit — for refractory skin disease.
- For calcinosis: no proven pharmacotherapy; surgical excision for symptomatic lesions. Trials of diltiazem, colchicine, and sodium thiosulfate with limited evidence.
Polymyositis
Polymyositis is an autoimmune myopathy characterised by subacute proximal muscle weakness without the cutaneous features of dermatomyositis. It is mediated by direct CD8+ cytotoxic T-cell attack on muscle fibres. PM is increasingly recognised as an entity that frequently overlaps with other connective tissue diseases (antisynthetase syndrome, scleroderma-overlap, mixed connective tissue disease).
Clinical Features
- Symmetrical proximal weakness developing over weeks to months.
- Dysphagia in 30–40% — may be presenting feature.
- No skin involvement (distinguishes from DM).
- May overlap with ILD, arthritis, Raynaud, mechanic's hands — screen for antisynthetase antibodies.
- Cardiac involvement (arrhythmia, conduction defects, myocarditis) in 10–30%.
Diagnosis
Requires: (1) proximal weakness, (2) elevated CK, (3) myopathic EMG with fibrillation potentials and short-duration polyphasic motor unit potentials, (4) muscle biopsy showing endomysial CD8+ T-cell infiltrate invading non-necrotic muscle fibres, and (5) absence of skin findings. Anti-Jo-1 (anti-histidyl-tRNA synthetase) is the most common associated antibody.
Treatment
Treatment mirrors dermatomyositis — high-dose prednisolone with early addition of steroid-sparing agent:
- First-line: Prednisolone 1 mg/kg/day + methotrexate (preferred) or azathioprine.
- Second-line: Mycophenolate mofetil, tacrolimus (especially for ILD), rituximab (refractory disease; PBS Authority Required for rheumatoid — off-label use).
- IVIg: 2 g/kg divided over 2–5 days for severe or refractory disease; PBS Authority Required via Specialist Drug Programs.
Inclusion Body Myositis
Sporadic inclusion body myositis (sIBM) is the most common acquired myopathy in patients over 50 years and the most common inflammatory myopathy in men. It is characterised by a combination of inflammatory and degenerative pathological features (rimmed vacuoles, amyloid deposits, TDP-43 aggregates). IBM is largely resistant to immunosuppressive therapy.
Clinical Features
- Insidious onset: Slowly progressive over years; often misdiagnosed as PM or age-related weakness.
- Finger flexor weakness: Weakness of flexor digitorum profundus (FDP) — cannot grip objects, turn keys, open jars. This pattern is highly characteristic.
- Quadriceps weakness: Early knee extensor weakness leading to falls and difficulty rising from seated position.
- Asymmetric involvement: Unlike DM/PM, IBM may be notably asymmetric.
- Dysphagia: Present in up to 60%; can be severe and life-threatening (aspiration risk).
- CK: Normal or only mildly elevated (typically <10× ULN).
Diagnosis
2011 ENMC criteria require: (1) duration >12 months, (2) age >45 years, (3) weakness of finger flexors and/or quadriceps, (4) CK <15× ULN, (5) EMG with myopathic or mixed pattern, and (6) muscle biopsy with endomysial inflammatory infiltrate AND rimmed vacuoles.
Management
- Physiotherapy: Supervised, progressive resistance training — maintains function, does not exacerbate disease.
- IVIg: May provide modest benefit in swallowing function; 2 g/kg over 2–5 days every 4 weeks for selected patients with significant dysphagia.
- Dysphagia: Speech pathology assessment, modified diet textures, oesophageal dilatation for cricopharyngeal dysfunction; consider botulinum toxin injection to upper oesophageal sphincter.
- Fall prevention: Home modifications, walking aids, quadriceps strengthening.
- Clinical trials: Emerging therapies — arimoclomol (heat shock protein co-inducer), sirolimus, rapamycin; refer to tertiary centres for trial access.
Immune-Mediated Necrotising Myopathy
Immune-mediated necrotising myopathy (IMNM), also termed necrotising autoimmune myopathy (NAM), is characterised by prominent myofibre necrosis with minimal inflammatory infiltrate on muscle biopsy. Two major autoantibody-defined forms exist: anti-HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase) and anti-SRP (signal recognition particle).
Anti-HMGCR Myopathy
- Strongly associated with statin exposure (atorvastatin, rosuvastatin most commonly in Australia), but also occurs statin-naïve, particularly in younger patients.
- Presents with severe proximal weakness and very high CK (often >10× ULN, frequently >3,000 U/L).
- Weakness persists and may worsen after statin cessation — distinguishes from self-limiting statin myopathy.
- Treatment: Immunosuppression as per DM/PM; often requires IVIg for refractory disease.
- Antibody titres correlate with disease activity — useful for monitoring.
Anti-SRP Myopathy
- Not associated with statins; presents with severe, rapidly progressive proximal weakness.
- Very high CK levels (often >5,000 U/L).
- Higher rates of cardiac involvement (arrhythmia, heart failure) — baseline ECG and echocardiogram recommended.
- Treatment: Often requires combination therapy — prednisolone + steroid-sparing agent + IVIg or rituximab.
- Generally more treatment-resistant than anti-HMGCR IMNM.
Myositis-Specific Antibodies
Myositis-specific antibodies (MSA) define distinct clinical phenotypes, predict cancer risk, inform screening urgency, and guide treatment. Testing is available at major Australian reference laboratories (including Royal Prince Alfred Hospital immunology, SA Pathology, and interstate referral centres).
| Antibody | Target | Subtype Association | Key Clinical Features | Cancer Risk |
|---|---|---|---|---|
| Anti-Mi-2 | Helicase | Classic DM | Classic skin disease, good treatment response | Low |
| Anti-TIF1-γ | Transcription intermediary factor | Adult DM | Prominent skin, cancer-associated myositis | High (~80%) |
| Anti-NXP2 | Nuclear matrix protein 2 | Adult DM, JDM | Calcinosis (JDM), muscle oedema, dysphagia | Moderate–High |
| Anti-MDA5 | Melanoma differentiation-associated protein 5 | Clinically amyopathic DM | Rapidly progressive ILD, skin ulceration, arthritis | Low (lung risk > cancer) |
| Anti-SAE | Small ubiquitin-like modifier activating enzyme | Adult DM | Dysphagia, skin disease before myopathy | Moderate |
| Anti-Jo-1 | Histidyl-tRNA synthetase | Antisynthetase syndrome (PM > DM) | ILD, arthritis, Raynaud, mechanic's hands, fever | Low |
| Anti-SRP | Signal recognition particle | IMNM | Severe necrotising myopathy, cardiac involvement | Low |
| Anti-HMGCR | HMG-CoA reductase | IMNM | Statin-associated or statin-naïve necrotising myopathy | Low |
Testing in Australia
- MSA panels available via line immunoassay (Euroline) or addressable laser bead immunoassay (ALBIA) at reference laboratories.
- MBS item: Not directly MBS-rebated as a panel; individual antibody tests may be billed under extended serology. Specialist referral required.
- Myositis-associated antibodies (MAA) — anti-PM-Scl, anti-Ku, anti-U1-RNP, anti-Ro52 — indicate overlap syndromes and should be tested in conjunction with MSA.
Cancer Screening in Adult Dermatomyositis
Adult-onset dermatomyositis carries a 4–7-fold increased risk of malignancy, with the highest risk in the first 3 years after diagnosis. Approximately 20–30% of adult DM patients will be diagnosed with cancer, most commonly within the first year. Cancer screening is therefore mandatory at diagnosis and must continue for at least 3–5 years.
Recommended Screening Protocol (Australian Practice)
- Full history and clinical examination (including breast, pelvic, testicular, thyroid, lymph node exam).
- CT chest, abdomen, and pelvis with contrast (MBS item 56809).
- Age-appropriate cancer screening: mammography (≥50 years; ≥40 with risk factors), cervical screening test (replaces Pap smear — 5-yearly from age 25), colonoscopy (≥50 years or per family history).
- Blood tests: FBC, LFTs, LDH, serum protein electrophoresis (SPEP), urinalysis with cytology.
- Consider: PET-CT if CT indeterminate or high clinical suspicion (MBS item 61306 — specialist referral).
- Consider: Upper GI endoscopy and colonoscopy if GI symptoms or age >50.
- Transvaginal ultrasound for ovarian assessment in women (not MBS-rebated routinely — discuss with patient).
- Repeat CT chest/abdomen/pelvis annually for the first 3 years (more frequently if TIF1-γ or NXP2 positive).
- Clinical vigilance at every rheumatology visit — new or worsening symptoms should prompt urgent re-investigation.
- Repeat age-appropriate screening per national guidelines.
- Myositis relapse or refractory disease should trigger repeat cancer screening.
- Anti-Mi-2 positive: Lower cancer risk; still perform baseline CT and age-appropriate screening but annual CT may not be required.
- Anti-MDA5 positive: Cancer risk is low; prioritise ILD screening (HRCT, PFTs).
- Juvenile DM: Cancer screening not routinely indicated.
Cancers Most Commonly Associated with DM
| Cancer Type | Relative Risk | Screening Method |
|---|---|---|
| Ovarian | Very high (up to 20×) | CT pelvis, transvaginal USS, CA-125 |
| Lung | High | CT chest |
| Pancreatic | High | CT abdomen |
| Gastric | Moderate–High | Upper GI endoscopy |
| Colorectal | Moderate | Colonoscopy (≥50 years) |
| Breast | Moderate | Mammography (biennial ≥50 years) |
| Lymphoma | Moderate | CT, PET-CT |
| Nasopharyngeal | Moderate (especially in SE Asian populations) | Nasopharyngoscopy, EBV serology |
Investigations
Risk Stratification & Severity Assessment
Severity assessment guides treatment intensity and monitoring frequency. Stratify patients at diagnosis based on functional impairment, organ involvement, antibody profile, and cancer risk.
Validated Outcome Measures
- MDAAT (Myositis Disease Activity Assessment Tool): Physician global activity (0–10 VAS), muscle VAS, extramuscular VAS.
- CMAS (Childhood Myositis Assessment Scale): 14-item functional scale; validated for children and useful in adults.
- MMT-8 (Manual Muscle Testing 8): Quantitative strength testing of 8 proximal muscle groups.
- HAQ-DI (Health Assessment Questionnaire — Disability Index): Patient-reported functional status.
Treatment — Empirical & Directed Therapy
First-Line Therapy (DM, PM, IMNM)
Second-Line & Refractory Disease
Allergy / Intolerance Alternatives
- Methotrexate intolerance (GI or hepatotoxicity): Switch to azathioprine or mycophenolate mofetil. Consider SC methotrexate to reduce GI side effects.
- Azathioprine (low TPMT): Switch to methotrexate or mycophenolate mofetil.
- Myositis-ILD (refractory to MMF): Add or switch to tacrolimus; consider rituximab.
- Corticosteroid-sparing in pregnancy: Azathioprine (safe), hydroxychloroquine (safe). Avoid methotrexate and mycophenolate (teratogenic).
Monitoring
- CK, aldolase, CRP, ESR, FBC, LFTs, renal function.
- Prednisolone dose and side effects (blood glucose, blood pressure, weight).
- Functional assessment: MMT-8, grip strength, chair rise, MDAAT.
- Methotrexate: FBC, LFTs, creatinine every 2 weeks until stable, then monthly.
- Azathioprine: FBC weekly for first month, then fortnightly, then monthly.
- CK and functional assessment — guide steroid tapering.
- Anti-HMGCR titres (where available) — correlate with disease activity.
- Pulmonary function (FVC, DLCO) every 6–12 months if ILD.
- DEXA scan annually while on corticosteroids (calcium + vitamin D supplementation).
- Ophthalmology review annually (hydroxychloroquine retinal toxicity if applicable).
- Annual CT chest/abdomen/pelvis for first 3 years (adult DM).
- Age-appropriate screening per national guidelines.
- Any change in disease pattern, new symptoms, or refractory disease — repeat cancer screening urgently.
Special Populations
Aboriginal and Torres Strait Islander Health
Inflammatory myopathies in Aboriginal and Torres Strait Islander peoples require culturally safe, accessible, and community-centred care. Limited data exist on the specific epidemiology of IIM in Indigenous Australians, but barriers to diagnosis and treatment are well-documented across autoimmune rheumatic diseases.
📚 References
- 1. Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 2017;69(12):2271–2282.
- 2. Allenbach Y, Mammen AL, Benveniste O, Stenzel W. 224th ENMC International Workshop: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies, Naarden, The Netherlands, 14–16 October 2016. Neuromuscul Disord. 2018;28(1):87–99.
- 3. Rose MR. 188th ENMC International Workshop: Inclusion body myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044–1055.
- 4. Tiniakou E,