📋 Key Information Summary
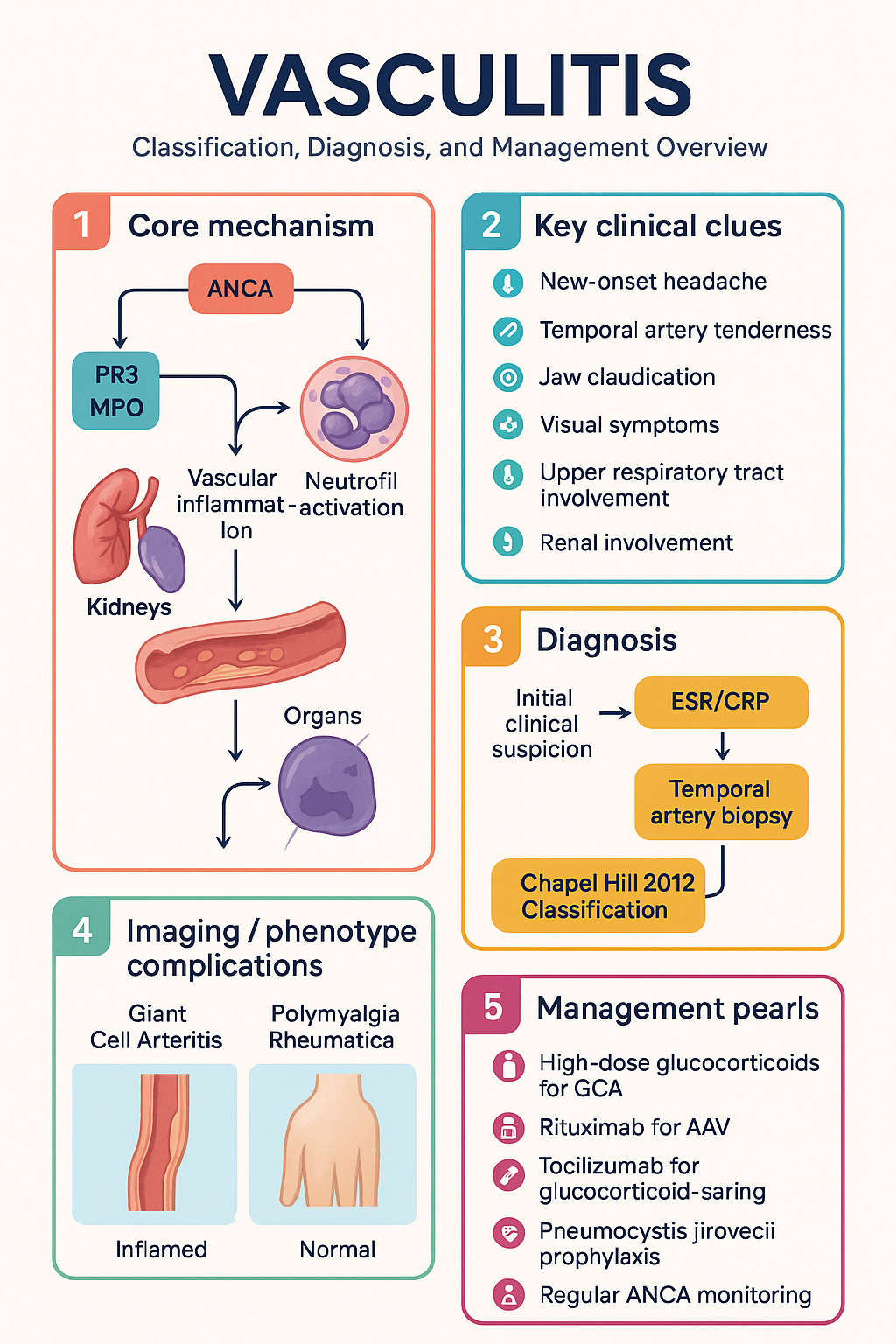
- The Chapel Hill 2012 Consensus Conference classifies vasculitis by predominant vessel size: large vessel (GCA, TAK), medium vessel (PAN, Kawasaki), and small vessel (ANCA-associated, immune-complex mediated)
- ANCA-associated vasculitis (AAV) comprises granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA); c-ANCA/PR3 is typical of GPA; p-ANCA/MPO is typical of MPA and EGPA
- Giant cell arteritis (GCA) is a medical emergency if vision is threatened — immediate high-dose IV methylprednisolone (500 mg–1 g daily for 3 days) is required before temporal artery biopsy
- Rituximab is non-inferior to cyclophosphamide for remission induction in severe AAV and is preferred as first-line for relapsing GPA; PBS Authority Required
- Tocilizumab is PBS-listed for giant cell arteritis and permits glucocorticoid-sparing in relapsing or refractory disease
- Avacopan (complement C5a receptor inhibitor) is TGA-approved as a glucocorticoid-sparing agent in severe AAV alongside standard induction therapy
- Polyarteritis nodosa (PAN) is associated with hepatitis B in ~30% of cases; antiviral therapy is essential alongside immunosuppression
- IgA vasculitis (Henoch–Schönlein purpura) is the most common childhood vasculitis; adults have higher risk of renal involvement and poorer outcomes
- All induction regimens for severe AAV require Pneumocystis jirovecii prophylaxis with trimethoprim–sulfamethoxazole (co-trimoxazole)
- BVAS (Birmingham Vasculitis Activity Score) is the standard tool for assessing disease activity in AAV
- Aboriginal and Torres Strait Islander peoples have higher rates of systemic inflammatory disease with later presentation and diagnostic delay
- Relapse prevention in AAV: azathioprine (PBS-listed) or rituximab maintenance for minimum 24 months; ongoing ANCA monitoring guides risk
- Always consider secondary causes of vasculitis — infection (HBV, HCV, HIV, endocarditis), drugs (hydralazine, propylthiouracil, allopurinol), and malignancy
Chapel Hill 2012 Classification
The 2012 International Chapel Hill Consensus Conference (CHCC) nomenclature is the current global standard for vasculitis classification. It stratifies primary vasculitides by the calibre of predominantly affected vessel and replaces the older 1994 classification.
| Vessel Size | Conditions | Key Features |
|---|---|---|
| Large vessel | Giant cell arteritis (GCA) Takayasu arteritis (TAK) |
Aorta and major branches; granulomatous inflammation; ischaemia of distal organs |
| Medium vessel | Polyarteritis nodosa (PAN) Kawasaki disease |
Mesenteric, renal, coronary arteries; necrotising inflammation without glomerulonephritis |
| Small vessel | ANCA-associated: GPA, MPA, EGPA Immune-complex: IgA vasculitis, cryoglobulinaemic vasculitis, anti-GBM disease |
Capillaries, venules, arterioles; glomerulonephritis, alveolar haemorrhage, purpura |
| Variable | Behçet disease, Cogan syndrome | Can affect vessels of any size |
The CHCC 2012 also introduced the category of "single-organ vasculitis" (e.g., cutaneous leukocytoclastic angiitis) and "vasculitis associated with systemic disease" or "probable aetiology" (e.g., HBV-associated PAN). These distinctions guide investigation for secondary causes.
ANCA-Associated Vasculitis (GPA, MPA, EGPA)
ANCA-associated vasculitis (AAV) encompasses three clinically overlapping but distinct entities: granulomatosis with polyangiitis (GPA, formerly Wegener's), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg–Strauss). Australian incidence is approximately 10–20 per million per year, with a peak in the 6th–7th decade.
Granulomatosis with Polyangiitis (GPA)
GPA classically presents with a triad of upper respiratory tract (sinusitis, saddle-nose deformity, subglottic stenosis), lower respiratory tract (nodules, cavitating lesions, alveolar haemorrhage), and renal involvement (pauci-immune necrotising glomerulonephritis). c-ANCA / anti-PR3 antibodies are positive in ~70–90%.
Microscopic Polyangiitis (MPA)
MPA typically presents with rapidly progressive glomerulonephritis (RPGN) and pulmonary capillaritis without granulomatous inflammation. p-ANCA / anti-MPO antibodies are positive in ~60–80%. MPA is the most common cause of pulmonary–renal syndrome in Australia.
Eosinophilic Granulomatosis with Polyangiitis (EGPA)
EGPA is characterised by asthma, peripheral eosinophilia (≥10% or ≥1.0 × 10⁹/L), migratory pulmonary infiltrates, neuropathy (mononeuritis multiplex), and vasculitis. p-ANCA/MPO positive in ~40% (ANCA-positive subgroup has higher renal involvement). The 2022 ACR/EULAR classification criteria incorporate eosinophil count, obstructive airway disease, nasal polyps, extravascular eosinophilic predominant inflammation, cytoplasmic ANCA or anti-MPO, and maximum eosinophil count ≥1.0 × 10⁹/L.
Severity Stratification
Induction Therapy
Glucocorticoid Regimen
Adjunctive Therapy
Maintenance Therapy
Refractory / Relapsing Disease
- Switch from cyclophosphamide to rituximab (or vice versa) if first-line induction fails
- Consider addition of plasma exchange (PLEX) — PEXIVAS trial showed no overall benefit for routine use, but may be considered for severe AKI (creatinine >500 µmol/L) or diffuse alveolar haemorrhage
- Mycophenolate mofetil (CellCept® 1–2 g/day BD) — PBS Restricted Benefit for lupus nephritis; off-label use in AAV with specialist approval
- Omalizumab may be considered for EGPA with refractory asthma (not PBS-listed for EGPA)
- Mepolizumab (Nucala®) 300 mg SC every 4 weeks — TGA-approved for EGPA; PBS Authority Required
Monitoring in AAV
Giant Cell Arteritis (GCA)
Giant cell arteritis is the most common primary systemic vasculitis in Australia, predominantly affecting adults >50 years (peak 70–80 years) with a female:male ratio of 2–3:1. Australian incidence is approximately 15–25 per 100,000 per year in those aged >50. GCA commonly coexists with polymyalgia rheumatica (PMR) in 40–60% of cases.
Clinical Presentation
- New-onset headache (usually temporal, may be diffuse) — most common symptom (~70%)
- Temporal artery abnormalities: tenderness, reduced pulsation, nodularity, beading
- Jaw claudication with chewing (~50%) — highly specific
- Visual symptoms: amaurosis fugax, diplopia, vision loss — emergency
- Constitutional: fever, weight loss, night sweats (may mimic sepsis or malignancy)
- Upper limb claudication (aortic arch / large-vessel GCA)
- PMR symptoms: bilateral shoulder and hip girdle stiffness >30 min
- Stroke or TIA (posterior circulation)
Investigations
Treatment
Without visual symptoms
With visual symptoms (amaurosis fugax / vision loss)
Glucocorticoid-Sparing Agents
Cardiovascular Risk in GCA
GCA patients have increased cardiovascular morbidity. Address modifiable risk factors: statin therapy, antihypertensives, smoking cessation, low-dose aspirin (75 mg daily — consider in all unless contraindicated). Monitor for aortic aneurysm with imaging at baseline and periodically.
Takayasu Arteritis
Takayasu arteritis (TAK) is a granulomatous large-vessel vasculitis predominantly affecting the aorta and its major branches. It typically presents before age 40 (often 15–30 years) with a strong female predominance (8–9:1). In Australia, TAK is more frequently seen in patients of Asian and Middle Eastern descent, though it affects all ethnicities.
Clinical Presentation
- Systemic phase: Fever, weight loss, arthralgia, myalgia — may be indolent for months
- Occlusive phase: Limb claudication, pulse deficits, bruits (subclavian, carotid, aortic), limb blood pressure discrepancy (>10 mmHg)
- Renovascular hypertension (renal artery stenosis)
- Aortic regurgitation, aortic root dilatation, coronary ostial stenosis
- Stroke/TIA (carotid/vertebral involvement)
- Pulmonary artery involvement (uncommon but recognised)
Diagnosis
Treatment
Polyarteritis Nodosa (PAN)
Polyarteritis nodosa is a necrotising medium-vessel vasculitis affecting muscular arteries. It spares the lungs and glomeruli (distinguishing it from AAV). Australian incidence is approximately 1–3 per million per year. Hepatitis B virus (HBV) is associated with ~30% of PAN cases worldwide; prevalence of HBV-PAN is lower in Australia due to vaccination programmes but remains relevant in unvaccinated populations.
Clinical Presentation
- Constitutional: fever, weight loss, malaise (almost universal)
- Cutaneous: livedo reticularis, nodules, purpura, digital gangrene, ulceration
- Renal: renovascular hypertension (arterial, not glomerular), renal infarction, microaneurysms on angiography
- Mesenteric: abdominal pain (post-prandial), bowel ischaemia/perforation, GI haemorrhage
- Neurological: mononeuritis multiplex (common), CNS vasculitis (rare)
- Musculoskeletal: myalgia, arthralgia (non-erosive)
- Testicular pain (orchitis)
- Coronary arteritis → myocardial infarction (rare)
Diagnosis
Treatment
HBV-Negative PAN
HBV-Associated PAN
- Short-course corticosteroids only (1–2 weeks) for symptom control — prolonged immunosuppression impairs viral clearance
- Antiviral therapy is the cornerstone: entecavir (Baraclude® 0.5 mg PO daily) or tenofovir (Viread® 300 mg PO daily) — PBS General Benefit
- Consider plasma exchange (PLEX) in severe cases
- Do NOT use rituximab or cyclophosphamide in HBV-PAN (impairs viral clearance)
- Monitor HBV DNA and LFTs; refer to hepatologist
IgA Vasculitis (Henoch–Schönlein Purpura)
IgA vasculitis (IgAV, formerly Henoch–Schönlein purpura) is the most common systemic vasculitis in children (peak age 3–10 years; 90% of cases are paediatric). Annual incidence in children is approximately 10–20 per 100,000. Adult IgAV is rarer but associated with more severe renal involvement. Characterised by IgA1-dominant immune complex deposition in small vessels.
Clinical Features (Classic Tetrad)
- Palpable purpura (100%): non-thrombocytopenic, gravity-dependent (buttocks, lower limbs); may be preceded by urticaria
- Arthritis / arthralgia (~75%): large joints (knees, ankles), non-erosive, migratory
- Abdominal pain (~65%): colicky, may precede purpura by days; risk of intussusception, bowel perforation, GI haemorrhage
- Nephritis (~30–50% in children, higher in adults): haematuria (microscopic or macroscopic), proteinuria, nephrotic syndrome, RPGN in severe cases
- Other: scrotal oedema, CNS vasculitis (rare), pulmonary haemorrhage (rare)
Diagnosis
Primarily clinical — EULAR/PRINTO/PRES 2010 criteria: palpable purpura (mandatory) with at least one of diffuse abdominal pain, biopsy showing predominant IgA deposition, arthritis/arthralgia, or renal involvement. No specific serological test is diagnostic; serum IgA may be elevated in 50% but is not required.
Treatment
Most cases are self-limiting in children (resolves within 4–6 weeks). Supportive care is the mainstay.
Severe Renal Disease
- Nephrotic syndrome or RPGN: high-dose prednisolone ± cyclophosphamide or mycophenolate mofetil (specialist nephrology management)
- ACE inhibitors for proteinuria control
- Refer to paediatric or adult nephrology — renal biopsy guides therapy
- Dapsone has been used for refractory skin disease (limited evidence)
Prognosis
Children: >95% have complete recovery. Relapse occurs in ~30% within 4 months. Adults: worse renal prognosis — up to 10–30% develop CKD or ESKD, particularly with crescentic nephritis on biopsy. Long-term nephrology follow-up is recommended for adults with any renal involvement.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
- 2. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis (RAVE trial). N Engl J Med. 2010;363(3):221–232.
- 3. Jones RB, Tervaert JWC, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis (RITUXVAS trial). N Engl J Med. 2010;363(3):211–220.
- 4. Walsh M, Merkel PA, Peh CA, et al. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis (PEXIVAS trial). N Engl J Med. 2020;382(7):622–631.
- 5. Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of maintenance therapy in ANCA-associated vasculitis (MAINRISAN trial). N Engl J Med. 2014;371(19):1771–1780.
- 6. Jayne DRW, Merkel PA, Schall TJ, Bekker P. Avacopan for the treatment of ANCA-associated vasculitis (ADVOCATE trial). N Engl J Med. 2021;384(7):599–609.
- 7. Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis (GiACTA trial). N Engl J Med. 2017;377(4):317–328.
- 8. Nakaoka Y, Isobe M, Takei S, et al. Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis (TAKT trial). JACC. 2018;71(14):1587–1596.
- 9. Lightfoot RW Jr, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. 1990;33(8):1088–1093.
- document.querySelectorAll('.topic-toc a[href^="#"]').forEach(o=>{o.addEventListener("click",i=>{const t=o.getAttribute("href")||"",e=document.querySelector(t);if(!e)return;i.preventDefault();const n=e.getBoundingClientRect().top+window.scrollY-70;window.scrollTo({top:n,behavior:"smooth"}),history.replaceState(null,"",t)})});const r=new Map;document.querySelectorAll(".topic-toc a").forEach(o=>{const i=o.getAttribute("href")?.slice(1);i&&r.set(i,o)});if(r.size>0&&"IntersectionObserver"in window){const o=new IntersectionObserver(i=>{let t=null;for(const e of i)if(e.isIntersecting){t=e.target.id;break}t&&r.forEach((e,n)=>e.classList.toggle("active",n===t))},{rootMargin:"-80px 0px -40% 0px",threshold:0});r.forEach((i,t)=>{const e=document.getElementById(t);e&&o.observe(e)})}window.matchMedia("(max-width: 900px)").matches&&document.querySelector(".topic-rail-collapsible")?.removeAttribute("open");const c=document.querySelector(".topic-rail-details");function s(){c&&(window.matchMedia("(max-width: 900px)").matches?c.open=!1:c.open=!0)}s();let l;window.addEventListener("resize",()=>{clearTimeout(l),l=window.setTimeout(s,120)});