📋 Key Information Summary
- AA amyloidosis results from sustained elevation of serum amyloid A (SAA) protein during chronic inflammatory conditions including rheumatoid arthritis, familial Mediterranean fever, juvenile idiopathic arthritis, and ankylosing spondylitis.
- AL amyloidosis arises from plasma cell dyscrasia (monoclonal immunoglobulin light chain deposition); associated with myeloma and monoclonal gammopathy of undetermined significance (MGUS).
- Congo red staining with apple-green birefringence under polarised light remains the histological gold standard for amyloid confirmation.
- AA amyloidosis primarily targets the kidney (nephrotic syndrome progressing to renal failure) and gastrointestinal tract (dysmotility, malabsorption).
- AL amyloidosis preferentially affects the heart (restrictive cardiomyopathy), kidneys, and peripheral/autonomic nerves.
- Macroglossia and periorbital purpura are pathognomonic clinical features of AL amyloidosis.
- Fat pad aspiration is the safest initial biopsy technique, with sensitivity of 60–80%; rectal biopsy supplements when negative.
- SAP scintigraphy (serum amyloid P component scan) quantifies whole-body amyloid burden but is available only at specialist centres in Australia.
- Cardiac MRI with late gadolinium enhancement pattern is the imaging modality of choice for cardiac amyloidosis.
- AA treatment centres on aggressive control of the underlying inflammatory disease: colchicine prevents AA in FMF; anti-IL-1 and anti-TNF agents target refractory inflammatory sources.
- AL treatment uses bortezomib/melphalan/dexamethasone (VMD) induction with consideration of autologous stem cell transplant (ASCT) in eligible patients.
- Prognosis in AA amyloidosis correlates with SAA levels and renal function; early suppression of inflammation halts amyloid deposition and may allow regression.
Introduction & Australian Epidemiology
Amyloidosis is a group of disorders characterised by extracellular deposition of misfolded protein in an abnormal fibrillar configuration. In the context of rheumatic disease, the two clinically relevant forms are AA (secondary) amyloidosis driven by chronic systemic inflammation, and AL (primary) amyloidosis caused by monoclonal immunoglobulin light chain deposition from plasma cell dyscrasia. Both lead to progressive organ dysfunction through disruption of normal tissue architecture and can be fatal if undiagnosed.
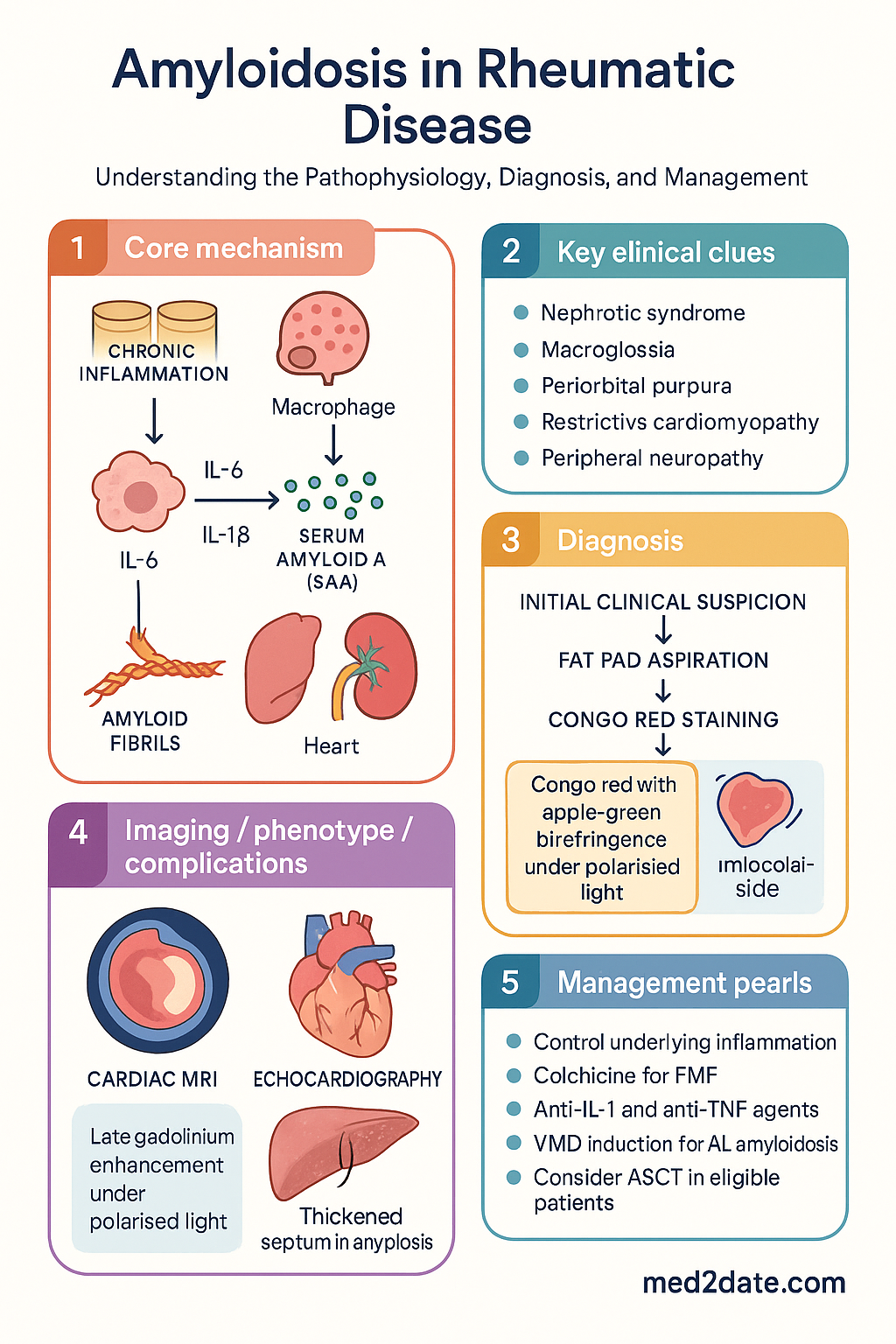
Secondary (AA) amyloidosis remains a serious systemic complication of inadequately controlled chronic inflammatory conditions, including rheumatoid arthritis (RA), familial Mediterranean fever (FMF), juvenile idiopathic arthritis (JIA), ankylosing spondylitis (AS), and Crohn disease. The unifying mechanism is sustained elevation of serum amyloid A (SAA), an acute-phase reactant produced by the liver under interleukin-6 (IL-6) and IL-1β stimulation.
In Australia, AA amyloidosis is rare but clinically significant. The AIHW reports that amyloidosis-related hospitalisations have a mortality rate exceeding 20% within 5 years of diagnosis. FMF is relatively prevalent among Australian communities of Middle Eastern and Mediterranean descent, particularly in Lebanese, Armenian, Turkish, and Sephardic Jewish populations. RA-related AA amyloidosis has declined markedly with early aggressive disease-modifying therapy but still occurs in patients with longstanding, poorly controlled disease. The prevalence of AL amyloidosis is estimated at 8–12 per million population per year in Australia.
Pathogenesis & Types
AA Amyloidosis (Secondary)
AA amyloidosis develops when persistent elevation of serum amyloid A (SAA) — an acute-phase apolipoprotein synthesised predominantly by hepatocytes — leads to proteolytic cleavage and misfolding into β-pleated amyloid fibrils. These fibrils deposit extracellularly in parenchymal organs and progressively replace normal tissue architecture.
The key pathological events are:
- Sustained inflammation: IL-6 and IL-1β drive hepatic SAA production. Chronic elevations (>10 mg/L persistently for months to years) are necessary but not sufficient for amyloid formation.
- Proteolytic processing: Monocyte-derived proteases cleave SAA into the N-terminal AA fragment, which is the primary constituent of AA fibrils.
- Fibril nucleation: AA fragments polymerise into cross-β fibrils. Glycosaminoglycans and serum amyloid P component (SAP) stabilise the fibril structure, resisting proteolysis.
- Organ deposition: Kidney is the predominant site (glomerular mesangium, basement membrane); liver, spleen, GI tract, and adrenal glands are also affected.
| Underlying Condition | Estimated Risk of AA | Typical Latency |
|---|---|---|
| Rheumatoid arthritis | 2–5% (historically higher) | 10–20 years of active disease |
| Familial Mediterranean fever | Up to 75% without colchicine | Childhood/adolescence |
| Juvenile idiopathic arthritis | 1–7% | 5–15 years |
| Ankylosing spondylitis | <1% | Variable, long-standing |
| Crohn disease | 0.5–1% | Variable |
| Other chronic infections (e.g., osteomyelitis, TB) | Case reports | Years of active infection |
AL Amyloidosis (Primary)
AL amyloidosis results from clonal expansion of plasma cells producing monoclonal immunoglobulin light chains (typically κ or λ) that misfold and deposit as amyloid fibrils. It is classified as a plasma cell dyscrasia and may coexist with myeloma, Waldenström macroglobulinaemia, or occur in the setting of MGUS. The light chain subtype determines organ tropism: AL deposits preferentially affect the heart, kidneys, nerves, and soft tissues.
Histological Diagnosis
All amyloid subtypes share a common histological appearance:
- Congo red staining: Amyloid deposits stain salmon-pink under conventional light microscopy.
- Apple-green birefringence under polarised light: This is the defining diagnostic feature and must be demonstrated for definitive amyloid confirmation.
- Subtyping: Immunohistochemistry (anti-AA antibody, anti-κ/λ antibodies) or mass spectrometry-based proteomics should be performed on all positive biopsies to determine the amyloid fibril type — critical for guiding therapy.
Clinical Manifestations
AA Amyloidosis
AA amyloidosis typically presents insidiously, with renal involvement as the dominant clinical feature. Patients often have long-standing inflammatory disease and may have proteinuria detected incidentally on urinalysis.
AL Amyloidosis
AL amyloidosis has multi-organ involvement and a broader clinical spectrum. It may mimic heart failure, nephrotic syndrome, neuropathy, or chronic diarrhoea — often simultaneously.
Diagnosis & Investigations
Biopsy Strategy
Tissue biopsy remains the cornerstone of amyloidosis diagnosis. The biopsy strategy depends on the clinical scenario and the likelihood of organ involvement.
Cardiac Assessment
Laboratory Investigations
| Test | Purpose | MBS Item / Notes |
|---|---|---|
| SAA level (serum amyloid A) | Monitor inflammatory activity in AA; target <4 mg/L to halt amyloid progression | Specialist pathology (reference lab) |
| Serum/urine protein electrophoresis + immunofixation | Detect monoclonal light chains in AL | MBS 66800/66803 |
| Serum free light chains (FLC) | Quantify κ/λ ratio; essential for AL diagnosis and response monitoring | MBS 66840 |
| NT-proBNP / BNP | Cardiac biomarker for AL staging and monitoring | MBS 66550 (BNP); NT-proBNP limited availability |
| High-sensitivity troponin | Prognostic staging in AL; predicts survival | MBS 66514 |
| 24-hour urine protein or spot protein:creatinine ratio | Quantify proteinuria and monitor renal response | MBS 66506 |
| eGFR, serum creatinine, serum albumin | Renal function and nutritional status | Routine MBS items |
| Bone marrow biopsy (for AL) | Clonal plasma cell burden; Congo red for marrow amyloid | Haematology referral |
Risk Stratification & Severity Scoring
AL Amyloidosis — Revised Mayo Clinic Staging
AL amyloidosis staging uses four prognostic biomarkers:
- NT-proBNP ≥1800 pg/mL
- hs-troponin T ≥0.025 ng/mL (or hs-troponin I elevated)
- Difference in free light chains (dFLC) ≥180 mg/L
AA Amyloidosis — Renal Prognostic Stratification
| Risk Category | eGFR at Diagnosis | Proteinuria | 5-Year Renal Survival |
|---|---|---|---|
| Low risk | ≥60 mL/min | <1 g/day | >85% |
| Intermediate risk | 30–59 mL/min | 1–5 g/day | 50–70% |
| High risk | <30 mL/min | >5 g/day | <30% |
Management — AA Amyloidosis
The fundamental principle in AA amyloidosis management is aggressive suppression of the underlying inflammatory disease to reduce SAA levels. The therapeutic target is sustained SAA <4 mg/L (ideally <2 mg/L). Amyloid regression can occur if SAA levels are controlled, but this requires months to years of sustained remission.
Disease-Modifying Therapy for Underlying Condition
Biologic DMARDs — Anti-TNF and Anti-IL-1
When conventional DMARDs fail to control inflammation, biologic agents targeting TNF-α or IL-1 are critical in suppressing SAA production.
Colchicine — FMF-Specific
Supportive Renal Care
- ACE inhibitors / ARBs: Recommended to reduce proteinuria and slow renal progression. Start at low dose and titrate cautiously (e.g., ramipril 2.5 mg daily → 10 mg daily).
- Diuretics: Loop diuretics for oedema management (furosemide 20–80 mg PO daily).
- Renal replacement therapy: Haemodialysis or peritoneal dialysis when eGFR <10 mL/min. Renal transplantation is an option but amyloid recurrence in the graft occurs in up to 30%.
- Antiplatelet agents: Consider in patients with nephrotic syndrome (thrombotic risk).
Management — AL Amyloidosis
AL amyloidosis management requires collaboration between haematology and the relevant organ specialists (cardiology, nephrology, neurology). The goals are eradication of the clonal plasma cell population, reduction of amyloidogenic light chain production, and supportive organ management.
First-Line Chemotherapy — Bortezomib/Melphalan/Dexamethasone (VMD)
Daratumumab-Based Regimens
Anti-CD38 monoclonal antibody daratumumab has transformed AL treatment. The ANDROMEDA trial demonstrated that daratumumab-bortezomib-melphalan-dexamethasone (D-VMD) achieved higher rates of complete haematological response and organ response compared to VMD alone. D-VMD is increasingly used as first-line in Australia.
Autologous Stem Cell Transplant (ASCT)
ASCT may be considered in selected patients with AL amyloidosis. Eligibility criteria are strict given the high transplant-related mortality (TRM) in patients with cardiac involvement.
Organ Support in AL
- Cardiac: Cautious diuretics (avoid hypotension); avoid digoxin and calcium channel blockers (bind to amyloid fibrils → toxicity); pacemaker/ICD for arrhythmias. Consider heart transplantation in selected patients with otherwise favourable prognosis.
- Renal: ACE inhibitors/ARBs for proteinuria; dialysis if needed. Renal transplantation with concurrent systemic therapy.
- Neuropathic: Neuropathic pain agents (gabapentin, pregabalin); physiotherapy for functional impairment; compression garments for autonomic instability.
- Nutritional: Dietitian input for malabsorption or macroglossia-related dysphagia.
Monitoring
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361–2371.
- 2. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38.
- 3. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541–4549.
- 4. Kastritis E, Leleu X, Arnulf B, et al. Bortezomib, melphalan, and dexamethasone with or without daratumumab for newly diagnosed multiple myeloma (ANDROMEDA): a phase 3, randomized, open-label trial. Lancet Oncol. 2021;22(12):1672–1684.
- 5. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360(23):2416–2425.
- 6. Bély M, Apáthy Á. Clinical pathology of rheumatoid arthritis: cause of death, lethal complications and associated diseases in rheumatoid arthritis. Acta Reumatol Port. 2012;37(2):101–122.
- 7. Robson KJ, Anstey AV, Barwell T, et al. Guidelines for the investigation and management of amyloidosis. British Committee for Standards in Haematology. 2015.
- 8. Pinney JH, Lachmann HJ. Systemic AA amyloidosis. Subcell Biochem. 2012;65:541–564.
- 9. Australian Institute of Health and Welfare (AIHW). Amyloidosis in Australia. Cat. no. PHE 312. Canberra: AIHW; 2023.
- 10. RHDAustralia (ARF/RHD writing group). The 2020 Australian guideline for prevention, diagnosis and management of acute rheumatic fever and rheumatic heart disease. 3rd edn. Menzies School of Health Research, Darwin; 2020.
- 11. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620–2627.
- 12. Lousada I, Comenzo RL, Landau H, et al. Light chain amyloidosis: patient experience survey from the Amyloidosis Research Consortium. Adv Ther. 2015;32(10):920–928.
- 13. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641–2654.
- 14. Fernández de Larrea C, Verga L, Morselli M, et al. The clinical value of mass spectrometry-based proteomics in amyloidosis diagnosis and prognosis. Haematologica. 2010;95(2):274–282.