📋 Key Information Summary
- Necrotising autoimmune myopathy (NAM) is a distinct inflammatory myopathy defined by myofibre necrosis with sparse lymphocytic inflammation, distinguished from dermatomyositis and polymyositis by serology and histopathology.
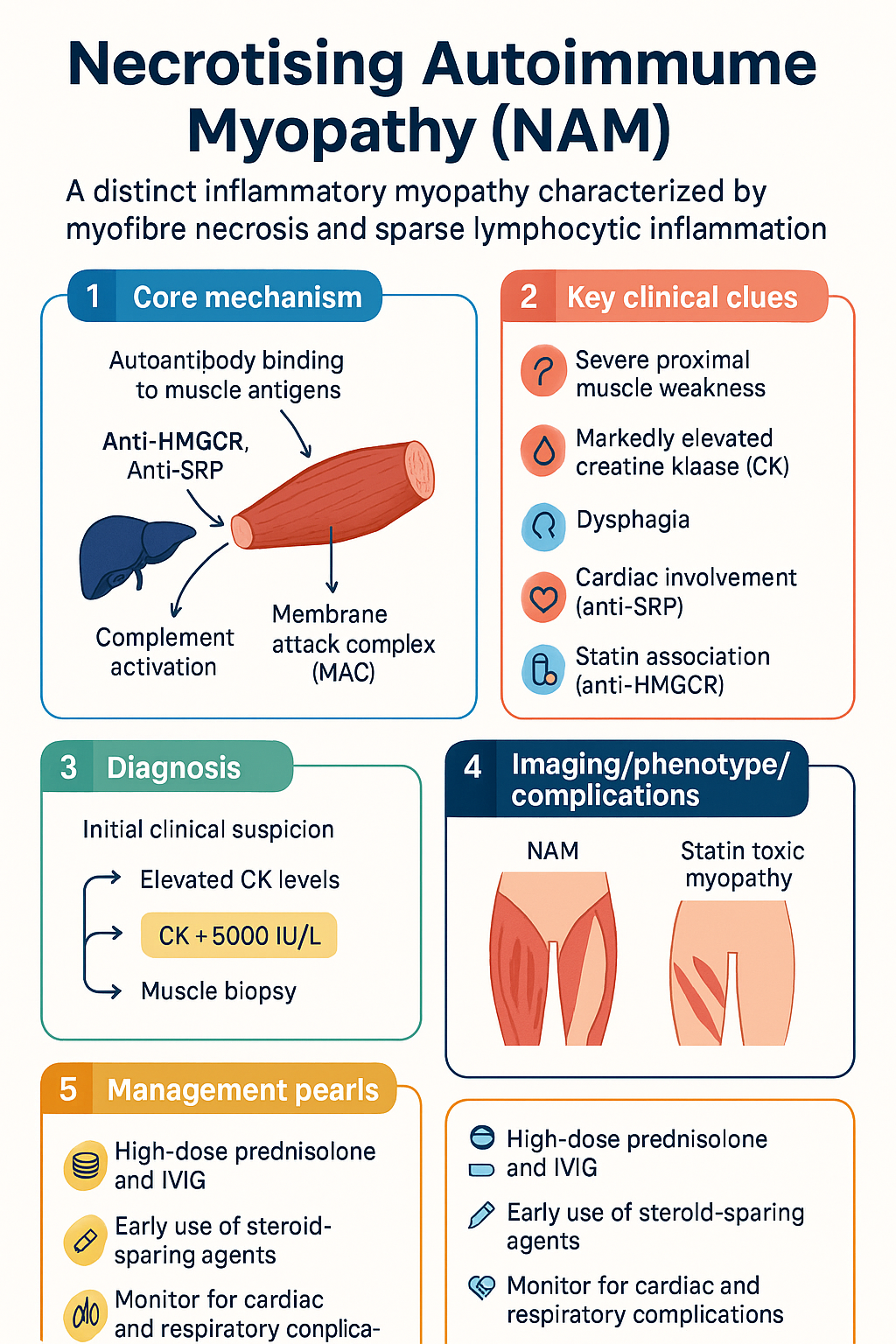
- Two major antibody subtypes: anti-HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase) — classically statin-associated; anti-SRP (signal recognition particle) — statin-independent and generally more severe.
- Anti-HMGCR NAM may occur in statin-exposed patients but also in statin-naïve individuals, particularly children and young adults of non-Caucasian descent.
- Anti-SRP NAM typically presents with severe, rapidly progressive proximal weakness and carries a higher risk of cardiac involvement and treatment resistance.
- Both antibody types activate complement-mediated myocyte injury via the membrane attack complex (MAC, C5b-9), leading to necrotic and regenerating fibres.
- Seronegative NAM exists and requires muscle biopsy for diagnosis — anti-HMGCR and anti-SRP serology alone is insufficient to exclude the diagnosis.
- Creatine kinase (CK) is typically markedly elevated, often >5000 IU/L and frequently >10,000 IU/L at presentation; CK serves as the primary treatment response marker.
- Muscle biopsy shows necrotic and regenerating fibres with minimal inflammatory infiltrate and MAC deposition on immunohistochemistry — unlike polymyositis where CD8+ T-cell infiltrates predominate.
- Discontinuation of statin alone is insufficient; aggressive combination immunotherapy is required in nearly all cases.
- First-line treatment: high-dose prednisolone 1 mg/kg/day (max 75 mg) PLUS intravenous immunoglobulin (IVIG) 2 g/kg over 2–5 days, then 1 g/kg monthly.
- Steroid-sparing agents: methotrexate (15–25 mg/week), azathioprine (2–3 mg/kg/day), or mycophenolate mofetil (2–3 g/day) should be commenced early; rituximab is effective for refractory disease in both subtypes.
- Treatment duration is prolonged — typically 2–5 years or longer; premature discontinuation carries a high relapse rate of 40–60%.
- Anti-SRP NAM may require more aggressive upfront therapy (e.g., early rituximab) due to higher rates of incomplete response and relapse compared with anti-HMGCR disease.
- Distinguish NAM from statin toxic myopathy (which resolves with statin cessation and does not require immunosuppression) — persistent CK elevation and weakness >2 months after statin withdrawal suggests NAM.
- Monitor for complications: dysphagia (assess with speech pathology), respiratory muscle weakness (FVC monitoring), cardiac involvement in anti-SRP (ECG, troponin, echocardiography), and steroid side effects including osteoporosis and glycaemic deterioration.
Introduction & Australian Epidemiology
Necrotising autoimmune myopathy (NAM) — also termed immune-mediated necrotising myopathy (IMNM) — is a relatively recently characterised inflammatory myopathy that has been formally distinguished from dermatomyositis, polymyositis, and inclusion body myositis in the 2017 European Neuromuscular Centre (ENMC) international classification. NAM is defined by a unique clinicopathological triad: severe proximal muscle weakness, markedly elevated serum creatine kinase (CK), and a muscle biopsy demonstrating prominent myofibre necrosis and regeneration with strikingly sparse lymphocytic inflammatory infiltrate.
The identification of disease-specific autoantibodies — anti-HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase) and anti-SRP (signal recognition particle) — has refined diagnostic accuracy and improved understanding of pathogenic mechanisms. These antibodies are now considered central to the diagnosis and subclassification of NAM, with seronegative cases representing approximately 10–20% of all presentations.
Epidemiology in Australia
NAM is a rare condition with an estimated annual incidence of 1–2 per million population globally. Australian-specific incidence data are limited, though extrapolation from international registries suggests approximately 25–50 new cases per year nationally. Key epidemiological observations in the Australian context include:
- Anti-HMGCR NAM is the most common subtype in Australia, consistent with the widespread use of statin therapy (approximately 2.7 million Australians are prescribed statins through the PBS).
- Anti-SRP NAM accounts for approximately 20–30% of NAM cases and appears to have no significant racial predilection in Australian cohorts.
- Statin-naïve anti-HMGCR NAM has been reported in children and young adults, with some evidence suggesting increased susceptibility in individuals of Southeast Asian descent — relevant given Australia's multicultural population.
- Anti-SRP NAM has been associated with cardiac involvement including myocarditis in up to 15–25% of cases, a complication with significant morbidity requiring specialist cardiology input.
- The condition affects adults across a wide age range (20–80 years), with anti-HMGCR cases peaking in the 5th–7th decades (correlating with statin exposure), while anti-SRP cases may present at younger ages.
Australia's universal healthcare system facilitates access to the specialist investigations required for NAM diagnosis (anti-HMGCR and anti-SRP serology available through major immunology laboratories, and PBS-subsidised access to IVIG and rituximab), though delays in diagnosis remain a significant concern — the median time from symptom onset to NAM diagnosis in published series is 3–6 months.
Pathogenesis & Antibodies
NAM is a humoural (antibody)-mediated myopathy in which autoantibodies directed against intracellular muscle antigens drive complement activation and myocyte necrosis. This contrasts with dermatomyositis (microangiopathy) and polymyositis (T-cell-mediated cytotoxicity). Two pathogenic antibody systems have been identified:
Anti-HMGCR Antibodies
- Anti-HMGCR antibodies are detected in approximately 60% of NAM cases in Australian tertiary referral series.
- The HMGCR enzyme is the pharmacological target of statin drugs (HMG-CoA reductase inhibitors).
- In statin-exposed patients, regenerating muscle fibres overexpress HMGCR, presenting an immunogenic target. Anti-HMGCR antibodies bind to surface-expressed HMGCR on regenerating fibres and activate the classical complement pathway, culminating in C5b-9 membrane attack complex (MAC) deposition and myofibre necrosis.
- Statin-naïve anti-HMGCR NAM occurs in approximately 30–40% of anti-HMGCR-positive patients, more commonly in children, young adults, and individuals of non-Caucasian ethnicity. The mechanism in this group is less well understood but may involve genetic polymorphisms in HMGCR expression regulation.
- HLA associations: HLA-DRB1*11:01 is strongly associated with anti-HMGCR NAM (odds ratio approximately 3.6), suggesting a genetic susceptibility component.
Anti-SRP Antibodies
- Anti-SRP antibodies target the 54 kDa subunit of the signal recognition particle, a ribonucleoprotein complex involved in co-translational protein translocation across the endoplasmic reticulum.
- Anti-SRP NAM is statin-independent — there is no established association between statin exposure and anti-SRP antibody development.
- Anti-SRP antibodies activate complement via both the classical and alternative pathways, leading to MAC deposition on the myocyte surface and necrosis.
- Anti-SRP NAM is generally considered more severe than anti-HMGCR disease, with higher presenting CK levels, more rapid progression, greater treatment resistance, and higher rates of extramuscular complications including cardiac involvement (myocarditis, conduction abnormalities).
- HLA associations: HLA-DRB1*08:03 has been identified in Japanese anti-SRP cohorts; the Australian HLA association is not well characterised.
Seronegative NAM
- Approximately 10–20% of patients with clinicopathological features consistent with NAM are negative for both anti-HMGCR and anti-SRP antibodies on standard testing.
- Seronegative NAM remains a histopathological diagnosis — the biopsy must demonstrate necrotic and regenerating fibres with minimal inflammation and MAC deposition to support the diagnosis.
- Emerging antibodies under investigation include anti-Mi2, anti-NXP2, and anti-SAE, though these are typically associated with dermatomyositis rather than pure NAM.
- Seronegative NAM should be managed with the same aggressive immunotherapy approach as seropositive disease.
Shared Pathogenic Mechanisms
Both anti-HMGCR and anti-SRP antibodies share a final common pathway of complement-mediated myocyte injury. Key mechanistic steps include:
- Autoantibody binding to intracellular target antigen (HMGCR on regenerating fibres surface; SRP on myocyte surface)
- Classical complement pathway activation (C1q binding to antibody Fc regions)
- C3 convertase formation and C3b amplification loop
- C5 convertase generation → C5a release (pro-inflammatory) → C5b-9 MAC assembly on myocyte membrane
- MAC pore formation → osmotic lysis → myofibre necrosis
- Satellite cell activation → regenerating fibres with HMGCR upregulation (perpetuating anti-HMGCR disease)
This complement-mediated mechanism provides the rationale for IVIG (complement scavenging and anti-idiotypic antibody effects) and rituximab (B-cell depletion reducing autoantibody production) as therapeutic strategies.
Clinical Features & Diagnosis
Clinical Presentation
NAM typically presents with subacute to chronic progressive proximal muscle weakness, developing over weeks to months. The clinical phenotype differs between the two major subtypes:
| Feature | Anti-HMGCR NAM | Anti-SRP NAM |
|---|---|---|
| Statin association | 60–70% statin-exposed; 30–40% statin-naïve | Statin-independent |
| Onset age | Typically 50–70 years (statin-associated); bimodal in children/young adults | Broad range 20–80 years; younger median age than anti-HMGCR |
| Weakness pattern | Symmetric proximal; may be subacute | Severe symmetric proximal; often rapidly progressive |
| Dysphagia | 20–30% | 30–50% |
| Cardiac involvement | Rare (<5%) | 15–25% (myocarditis, conduction defects) |
| ILD | Uncommon | Reported in up to 10–15% |
| Skin findings | None (distinguishes from dermatomyositis) | None |
| Treatment response | Generally better; 70–80% achieve remission | More refractory; 50–60% achieve complete response |
Characteristic Features
- Proximal weakness: Difficulty rising from chairs, climbing stairs, lifting arms above head, combing hair. Proximal upper and lower limbs are affected symmetrically.
- Severity: Weakness is often severe at presentation — many patients have significant functional impairment (MRC grade 3–4/5 in proximal muscles).
- Dysphagia: Occurs in 20–50% of cases; may be the presenting feature; carries aspiration risk — assess with speech pathology referral.
- Fatigue: Profound and often disproportionate to the degree of weakness.
- Myalgia: Present in approximately 30–50% but is not a predominant feature; patients more commonly report weakness than pain.
- Respiratory involvement: Respiratory muscle weakness (diaphragm, intercostals) may occur, particularly in anti-SRP NAM; monitor forced vital capacity (FVC).
- No skin involvement: The absence of rash (heliotrope, Gottron's papules, mechanic's hands) distinguishes NAM from dermatomyositis and is a critical diagnostic feature.
Diagnostic Criteria
Differential Diagnosis
| Condition | Key Distinguishing Features |
|---|---|
| Statin toxic myopathy | Resolves with statin cessation within 4–6 weeks; CK normalises; no autoantibodies; does NOT require immunosuppression |
| Dermatomyositis | Rash present (heliotrope, Gottron's); perifascicular atrophy on biopsy; anti-Mi2, anti-MDA5, anti-NXP2, anti-TIF1γ antibodies |
| Polymyositis | CD8+ T-cell endomysial infiltrates on biopsy; anti-Jo1 (and other antisynthetase) antibodies |
| Inclusion body myositis | Asymmetric; finger flexor/wrist flexor/quadriceps predilection; rimmed vacuoles on biopsy; poor treatment response; older males |
| Muscular dystrophy | Family history; slowly progressive; genetic testing diagnostic; biopsy may show dystrophic changes |
| Endocrine myopathy | Thyroid, adrenal, parathyroid disease; CK usually mildly elevated; responds to endocrine correction |
| Drug/toxin myopathy | Colchicine, chloroquine, alcohol, cocaine; temporal relationship; resolves with drug withdrawal |
| Viral myositis | HIV, HCV, influenza; acute onset; viral serology positive |
Investigations
Laboratory Investigations
Treatment
Treatment Principles
- Combination immunotherapy is required — monotherapy with corticosteroids alone has a high relapse rate.
- Initiate treatment early — delay correlates with irreversible muscle damage and fibrosis.
- Anti-SRP NAM generally requires more aggressive upfront therapy than anti-HMGCR disease.
- CK normalisation is the primary treatment target; complete clinical remission (normal strength + normal CK) is the long-term goal.
- Treatment duration is prolonged — minimum 2 years of sustained remission before cautious tapering; many patients require treatment for 5+ years.
- Premature discontinuation carries a 40–60% relapse rate.
First-Line Induction Therapy
Steroid-Sparing Agents (Commence within 4–8 Weeks)
A steroid-sparing agent should be started early to facilitate corticosteroid tapering and reduce cumulative steroid toxicity. Choice depends on patient factors including reproductive plans, hepatic and renal function, and comorbidities.
Second-Line / Refractory Disease — Rituximab
Treatment Response Monitoring
Special Populations
ATSI Health Considerations
📚 References
- 1. Allenbach Y, Mammen AL, Benveniste O, Stenzel W. 224th ENMC International Workshop: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscul Disord. 2018;28(1):87–99.
- 2. Allenbach Y, Drouot L, Rigolet A, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore). 2014;93(3):150–157.
- 3. Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry. 2016;87(10):1038–1044.
- 4. Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63(3):713–721.
- 5. Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol. 2015;72(9):996–1003.
- 6. Pinal-Fernandez I, Parks C, Werner JL, et al. Longitudinal profiling of autoantibodies in anti-HMGCR immune-mediated necrotizing myopathy. Neurology. 2017;88(6):574–581.
- 7. Suzuki S, Nishikawa A, Kuwana M, et al. Inflammatory myopathy with anti-signal recognition particle antibodies: a nationwide survey in Japan. Arthritis Res Ther. 2020;22(1):20.
- 8. Australian Commission on Safety and Quality in Health Care (ACSQHC). National Safety and Quality Health Service Standards. 2nd ed. Sydney: ACSQHC; 2021.
- 9. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2020 summary report. Canberra: AIHW; 2020.
- 10. Limaye V, Lester S, Blumbergs P, Greenberg SA. Idiopathic inflammatory myopathy — a review of classification, clinical features and treatment. Intern Med J. 2012;42(3):258–269.
- 11. Rider LG, Aggarwal R, Pistorio A, et al. 2016 American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis. Ann Rheum Dis. 2017;76(5):792–801.
- 12. The Royal Australian College of General Practitioners (RACGP). National guide to a preventive health assessment for Aboriginal and Torres Strait Islander people. 3rd ed. East Melbourne: RACGP; 2018.
- 13. Raval NA, Bhagat K, Stenzel W, Benveniste O, Allenbach Y. Auto-immune necrotizing myopathy: updates on clinical, diagnostic and therapeutic approaches. Curr Opin Rheumatol. 2023;35(5):299–308.
- 14. Dalakas MC. Inflammatory muscle diseases: a review of pathogenesis and treatment. Nat Rev Rheumatol. 2023;19(7):411–425.
- 15. Services Australia. Lifeblood (Australian Red Cross Blood Service) National IVIG Guidelines — clinical criteria for IVIG use in autoimmune neurological and rheumatological conditions. Canberra: Australian Government Department of Health; 2023.