📋 Key Information Summary
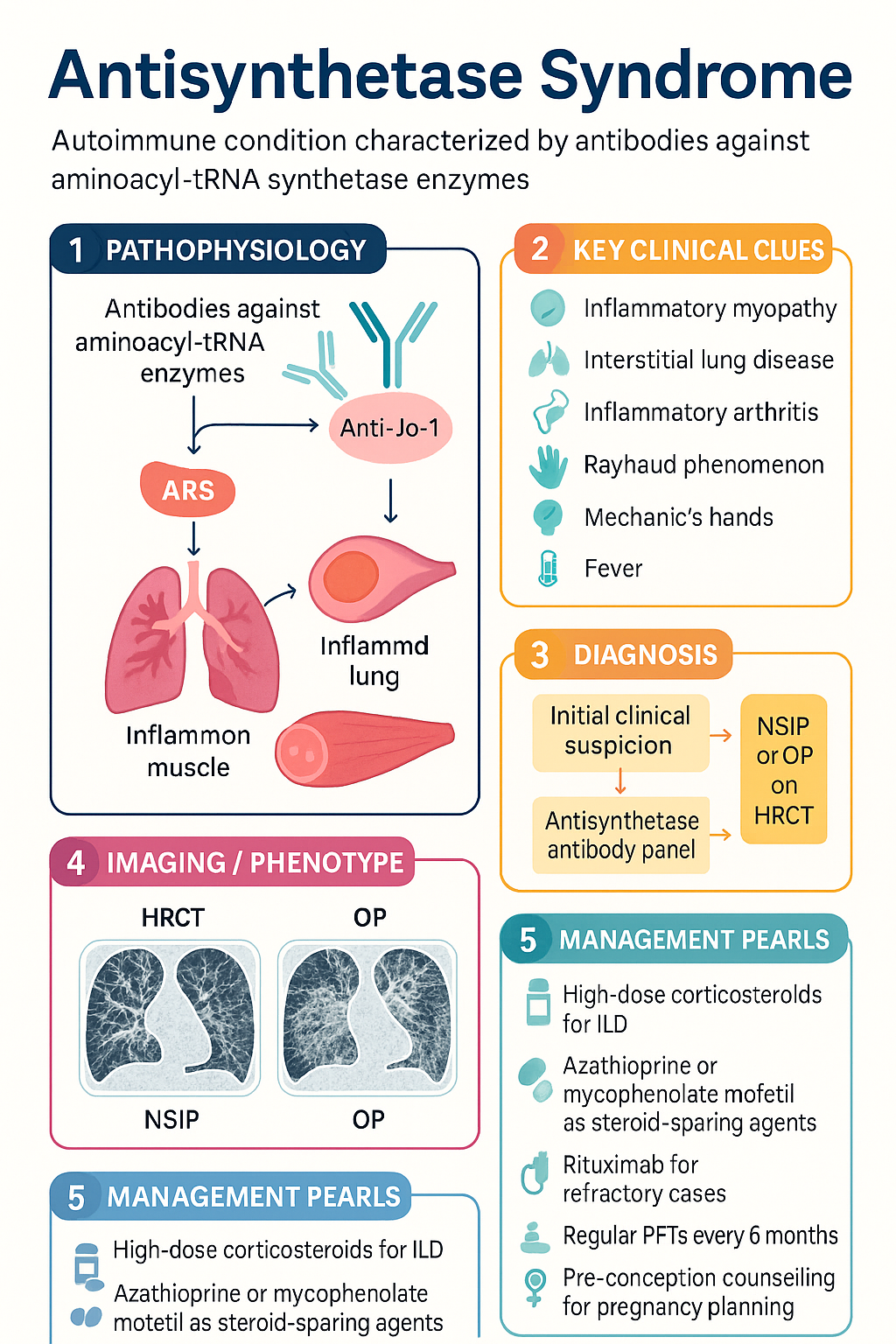
- Antisynthetase syndrome (ASS) is an autoimmune condition defined by antibodies against aminoacyl-tRNA synthetase enzymes — most commonly anti-Jo-1 (histidyl-tRNA synthetase), followed by anti-PL-7, anti-PL-12, anti-EJ, and anti-OJ.
- The classic six-component clinical cluster comprises inflammatory myopathy, interstitial lung disease (ILD), inflammatory arthritis, Raynaud phenomenon, mechanic's hands (hyperkeratotic fissuring), and fever.
- ILD is often the most clinically significant and prognostically important manifestation, frequently presenting as non-specific interstitial pneumonia (NSIP) or organising pneumonia (OP) pattern on HRCT.
- Anti-Jo-1–positive patients tend to have the most complete syndrome (myositis + ILD + arthritis); anti-PL-12– and anti-PL-7–positive patients may present with ILD-dominant disease and minimal myositis.
- Diagnosis requires a positive antisynthetase antibody plus at least one clinical feature — confirmation relies on HRCT chest, PFTs (FVC, DLCO), CK/aldolase, muscle MRI, EMG, and/or muscle biopsy.
- First-line ILD therapy: high-dose corticosteroids with a steroid-sparing agent — azathioprine or mycophenolate mofetil.
- Refractory or severe ILD/myositis: rituximab (preferred biologic); cyclophosphamide for rapidly progressive ILD; IVIg for refractory myositis.
- Pulmonary function testing (FVC, DLCO) every 6 months is mandatory to detect ILD progression; HRCT at baseline and when clinically indicated.
- Pregnancy planning requires pre-conception counselling — mycophenolate and methotrexate are teratogenic and must be ceased ≥6 weeks and ≥3 months pre-conception respectively; azathioprine and hydroxychloroquine are preferred maintenance agents.
- Screen for pulmonary arterial hypertension (PAH) with echocardiography; antinuclear antibody (ANA) may be negative — do not exclude ASS on ANA negativity alone.
- Aboriginal and Torres Strait Islander peoples may have higher rates of ILD and face barriers to specialist rheumatology and respiratory access — early referral and culturally safe care pathways are essential.
- Mortality is driven primarily by respiratory failure from progressive ILD; early aggressive treatment of lung disease improves outcomes.
Introduction & Australian Epidemiology
Antisynthetase syndrome (ASS) is a distinct autoimmune connective tissue disease characterised by the presence of autoantibodies directed against aminoacyl-tRNA synthetase enzymes, coupled with a constellation of clinical features including inflammatory myopathy, interstitial lung disease (ILD), inflammatory arthritis, Raynaud phenomenon, mechanic's hands, and fever. First described in the 1970s–1980s in patients with polymyositis and anti-Jo-1 antibodies, ASS is now recognised as a syndrome in its own right rather than a subset of dermatomyositis or polymyositis.
ASS accounts for approximately 30–40 % of all patients with idiopathic inflammatory myopathies (IIM) in Australian referral centre cohorts. The estimated prevalence in Australia is 1–2 per 100,000 population, with a female predominance (approximately 2:1 F:M). Peak incidence occurs in the 4th–6th decade of life, though ASS can present at any age including in paediatric populations.
Eight antisynthetase antibodies have been identified. Anti-Jo-1 (anti-histidyl-tRNA synthetase) is the most common, found in approximately 20–30 % of IIM patients and accounting for ~75 % of ASS cases. Other antibodies include anti-PL-7 (threonyl), anti-PL-12 (alanyl), anti-EJ (glycyl), anti-OJ (isoleucyl), anti-KS (asparaginyl), anti-Ha (tyrosyl), and anti-Zo (phenylalanyl). Each antibody defines a clinically distinct ASS phenotype — anti-PL-12 and anti-PL-7 patients more frequently present with ILD as the dominant or sole manifestation and may lack significant muscle disease.
In Australia, ILD is the leading cause of morbidity and mortality in ASS. Australian registry data indicate that approximately 70–90 % of ASS patients have evidence of ILD on HRCT at presentation, with NSIP and OP being the predominant radiological and histopathological patterns. Early recognition and treatment of ILD is therefore critical to improving long-term outcomes.
Clinical Features — The Six-Component Cluster
ASS is defined by the presence of at least one antisynthetase antibody plus at least one of the following six clinical features. Anti-Jo-1–positive patients most commonly exhibit the complete sextet, while other antibody subsets may present with fewer components.
| Feature | Prevalence | Key Details |
|---|---|---|
| 1. Inflammatory myopathy | 60–80 % | Proximal muscle weakness (shoulder/hip girdle); CK often elevated (may be normal in anti-PL-12); may be subtle or subclinical with EMG/MRI abnormalities only |
| 2. Interstitial lung disease | 70–90 % | NSIP (most common), OP, or UIP pattern on HRCT; progressive dyspnoea, dry cough; may precede myositis by months–years; dominant cause of mortality |
| 3. Inflammatory arthritis | 50–70 % | Symmetric, non-erosive polyarthritis; small joints of hands, wrists, knees; may mimic RA; RF and anti-CCP usually negative |
| 4. Mechanic's hands | 40–60 % | Hyperkeratotic, fissuring of lateral digits; rough, cracked skin with dyschromia; pathognomonic finding in ASS |
| 5. Raynaud phenomenon | 40–60 % | Episodic digital ischaemia — white → blue → red sequence; may antedate other features; less severe than in SSc |
| 6. Fever | 20–40 % | Often low-grade; may be the presenting feature; exclude infection in immunosuppressed patients |
Antibody-Specific Phenotypes
| Antibody | Frequency | Predominant Phenotype |
|---|---|---|
| Anti-Jo-1 | ~75 % of ASS | Most complete syndrome: myositis + ILD + arthritis + mechanic's hands |
| Anti-PL-7 | ~5–10 % | ILD-dominant; lower CK; higher mortality from progressive ILD |
| Anti-PL-12 | ~5–10 % | ILD-dominant; myositis often subtle or absent; worse ILD prognosis |
| Anti-EJ | ~3 % | Myositis + ILD; overlap features |
| Anti-OJ | ~1–2 % | Rare; ILD + myositis; often incomplete syndrome |
| Anti-KS, anti-Ha, anti-Zo | Rare | Limited data; ILD reported |
Pathophysiology
Aminoacyl-tRNA synthetases (ARS) are ubiquitous intracellular enzymes essential for protein synthesis, catalysing the attachment of amino acids to their cognate tRNA molecules. In ASS, autoantibodies target specific ARS enzymes. The pathogenic mechanisms linking these antibodies to tissue injury remain incompletely understood, but several mechanisms have been proposed:
- Direct antigen-driven immune activation: ARS enzymes may be expressed on the cell surface under conditions of cellular stress (e.g., viral infection, tissue injury), becoming accessible to autoantibodies and triggering complement-mediated and antibody-dependent cellular cytotoxicity.
- T-cell–mediated muscle injury: CD8+ cytotoxic T cells infiltrate perimysial and endomysial regions of skeletal muscle, driving perifascicular atrophy and myofibre necrosis — a pattern distinct from dermatomyositis (microangiopathy) and overlap myositis.
- Pulmonary fibrosis driven by TGF-β and pro-fibrotic cytokines: In the lung, chronic inflammation of the alveolar epithelium and peribronchiolar interstitium leads to fibroblast activation, collagen deposition, and progressive fibrosis — particularly in NSIP and UIP patterns.
- Molecular mimicry: Structural homology between ARS epitopes and viral proteins may initiate cross-reactive immune responses, consistent with epidemiological observations of viral triggers.
- Genetic susceptibility: HLA-DRB1*03:01 and HLA-DQA1*05:01 are strongly associated with anti-Jo-1 ASS in Caucasian populations; HLA-B*35 and HLA-DRB1*11:01 have been linked to anti-PL-12 ILD-dominant disease.
Diagnosis
Diagnosis of ASS requires the presence of a positive antisynthetase antibody plus at least one clinical feature from the six-component cluster. No single international classification criteria has been universally adopted; however, the 2010 Connors criteria and the emerging EULAR/ACR classification for IIM (2017) are commonly applied in Australian practice.
Diagnostic Approach
Investigations
Laboratory Investigations
Imaging
Electrodiagnostic & Histopathological
Risk Stratification & Prognosis
Prognosis in ASS is determined primarily by the severity and progression of ILD. Myositis-related mortality is low with modern immunosuppressive therapy; however, progressive pulmonary fibrosis accounts for the majority of disease-related deaths.
Management — Immunosuppressive Therapy
Treatment of ASS requires a dual approach targeting both the myositis and ILD components. ILD-directed therapy is the priority in most patients, as progressive pulmonary fibrosis drives mortality. All treatment decisions should be made in conjunction with rheumatology and respiratory medicine.
First-Line Induction Therapy
Second-Line / Refractory Disease
Other Agents — Limited Evidence
Monitoring
Structured monitoring is essential to detect ILD progression, myositis relapse, and medication toxicity. All ASS patients require long-term follow-up with rheumatology and respiratory medicine (pulmonology).
Monitoring Schedule
PFT Decline Thresholds — Action Points
| Change | Action |
|---|---|
| FVC decline ≥10 % relative (or ≥5 % absolute) | Repeat HRCT; consider treatment escalation (add/change steroid-sparing agent); multidisciplinary ILD team review |
| FVC decline ≥10 % in 3 months or ≥20 % in 6 months | Rapidly progressive ILD — consider cyclophosphamide or rituximab; urgent respiratory/rheumatology review; lung transplant referral |
| DLCO <40 % predicted | High mortality risk; evaluate for PAH (right heart catheterisation); lung transplant assessment |
Special Populations
ATSI Health Considerations
📚 References
- 1. Connors GR, Christopher-Stine L, Cosgrove O, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. 2010;138(6):1464–1474.
- 2. Lega JC, Fabien N, Reynaud Q, et al. The clinical phenotype associated with myositis-specific and associated antibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev. 2014;13(9):883–891.
- 3. Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 2017;69(12):2271–2282.
- 4. Hervier B, Devilliers H, Stanciu R, et al. Hierarchical clinical and serological classification of patients with anti-synthetase syndrome. Medicine (Baltimore). 2012;91(5):236–244.
- 5. Tieu J, Lundberg IE, Limaye V. Idiopathic inflammatory myositis. Best Pract Res Clin Rheumatol. 2022;36(1):101746.
- 6. Baccaro AC, Ceribelli A, Aoki MN, et al. Antisynthetase syndrome: clinical features and outcomes of a Brazilian cohort. Adv Rheumatol. 2020;60(1):53.
- 7. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–2666. [Scleroderma Lung Study — extrapolated to ASS-ILD]
- 8. Tieu J, Giri N, Limaye V. Management of interstitial lung disease in antisynthetase syndrome: an update. Curr Treat Options Rheumatol. 2021;7:244–259.
- 9. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2023 summary report. Canberra: AIHW; 2023.
- 10. Hallowell R, Danoff SK. Interstitial lung disease associated with the antisynthetase syndrome. J Clin Med. 2023;12(4):1391.
- 11. Allenbach Y, Uzunhan Y, Guenou H, et al. Different phenotypes in dermatomyositis associated with anti-Mi2 antibody. Neurology. 2020;94(1):e71–e80.
- 12. Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice (Red Book). 10th ed. East Melbourne: RACGP; 2023.
- 13. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47.
- 14. Australian Technical Advisory Group on Immunisation (ATAGI). Australian immunisation handbook. Canberra: Australian Government Department of Health; 2023. Available from: immunisationhandbook.health.gov.au.