📋 Key Information Summary
- Most common acquired inflammatory myopathy in adults over 50 years; prevalence ~50–70 per million in those aged >50 years, with increasing incidence in ageing populations.
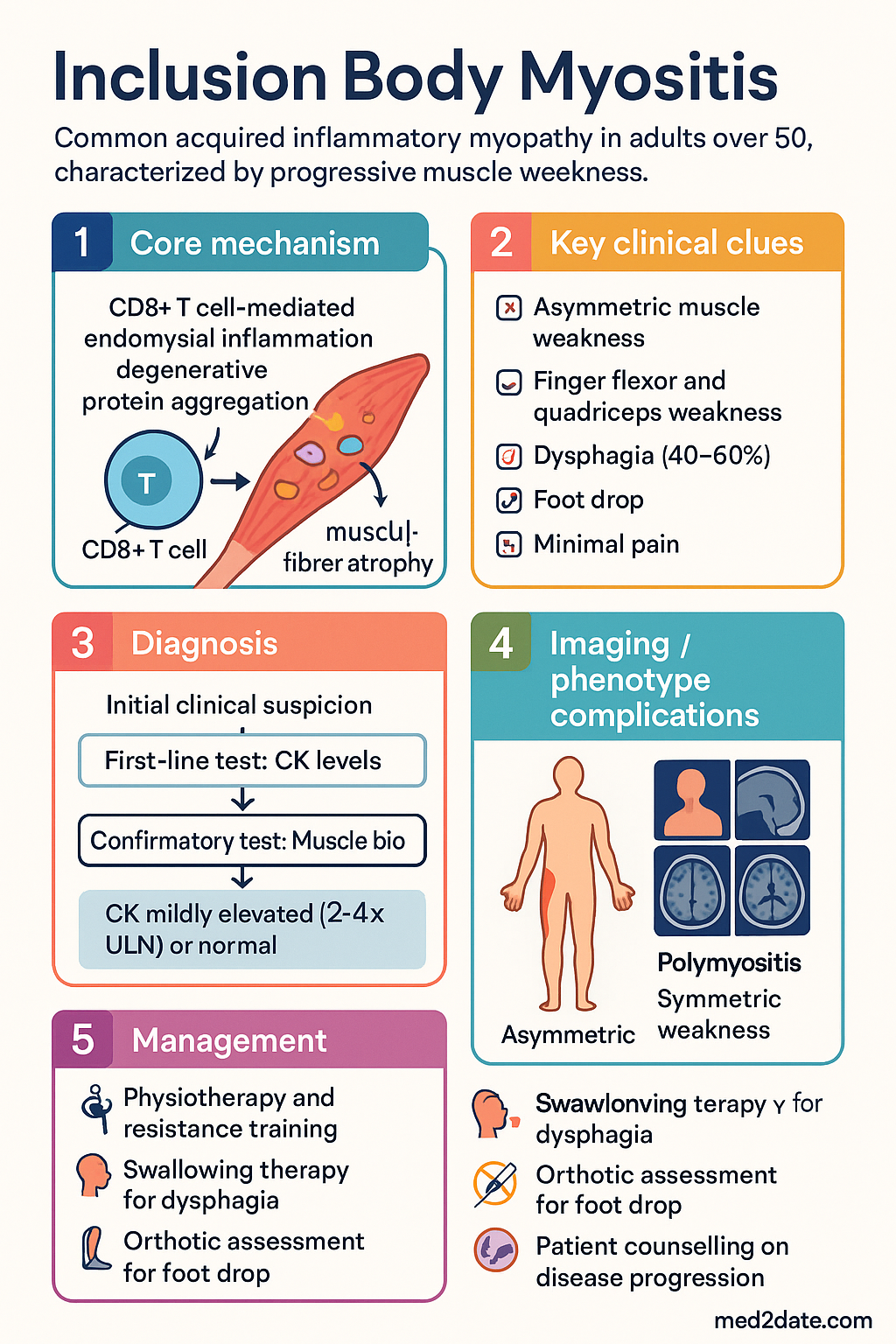
- Dual pathogenesis: degenerative protein aggregation (p62, TDP-43, amyloid-β, phosphorylated tau) combined with CD8+ T-cell-mediated endomysial inflammation — neither pathway alone explains the disease.
- Distinctive clinical pattern: slowly progressive, asymmetric weakness affecting finger flexors (grip strength, difficulty opening jars, pinch weakness) and quadriceps (difficulty rising from chairs, climbing stairs, frequent falls).
- Dysphagia occurs in 40–60% of patients and may predate limb weakness; it is the most dangerous complication due to aspiration risk.
- Foot drop from ankle dorsiflexor weakness increases fall risk and necessitates early orthotic assessment.
- Anti-cN-1A (NT5C1A) antibodies present in 30–60% — moderate sensitivity but helpful when positive; do not exclude IBM if negative.
- CK is often only mildly elevated (2–4× upper limit of normal) or even normal, which frequently delays diagnosis.
- Muscle biopsy is the gold standard: endomysial CD8+ T-cell infiltration, rimmed vacuoles, COX-negative fibres, and p62/TDP-43 immunoreactive inclusions.
- No proven disease-modifying therapy: IBM is largely refractory to corticosteroids and conventional immunosuppressants — an important distinction from polymyositis and dermatomyositis.
- Physiotherapy is the mainstay of management; progressive resistance training is safe and may modestly slow functional decline.
- Swallowing therapy and PEG tube consideration for severe or progressive dysphagia to prevent aspiration pneumonia.
- Prognosis: most patients require walking aids within 10–15 years; approximately 30% are wheelchair-dependent at 10 years from diagnosis.
- ATSI populations: limited data but equitable access to specialist neuromuscular services, NDIS, and allied health must be ensured.
Introduction & Australian Epidemiology
Sporadic inclusion body myositis (IBM) is the most common acquired inflammatory myopathy in adults aged over 50 years and is increasingly recognised as one of the leading causes of progressive muscle weakness in the elderly population worldwide. In Australia, IBM poses a growing clinical burden as the population ages, with estimated prevalence figures of 50–70 per million in those over 50 years rising to over 100 per million in those over 70 years.
IBM has a male-to-female ratio of approximately 2–3 : 1 and typically presents after the fifth decade, though earlier-onset cases are well described. The disease is characterised by slowly progressive, often asymmetric muscle weakness with a distinctive predilection for the quadriceps (knee extensors) and the deep finger flexors (flexor digitorum profundus). This pattern distinguishes IBM from other inflammatory myopathies — polymyositis (PM) and dermatomyositis (DM) — which tend to affect proximal, symmetric muscle groups.
Diagnosis is frequently delayed by 5–7 years because CK levels are often only mildly elevated or normal, and the clinical pattern may be misattributed to osteoarthritis, neuropathy, or age-related sarcopenia. In Australian tertiary neuromuscular centres, IBM accounts for approximately 16–30% of all inflammatory myopathy referrals, reflecting both true prevalence and growing clinician awareness.
A critical feature of IBM is its poor or absent response to immunosuppressive therapies. Unlike PM and DM, which generally respond to corticosteroids and steroid-sparing agents, IBM is largely refractory to such treatment. This has major implications for patient counselling and resource allocation, shifting the emphasis from pharmacological immunosuppression towards rehabilitation, symptom management, and multidisciplinary supportive care.
Pathogenesis & Clinical Features
Dual Pathogenic Pathways
The pathogenesis of IBM is incompletely understood but is best conceptualised as a convergence of two overlapping pathological processes: an inflammatory/autoimmune component and a degenerative/protein-aggregation component. Neither pathway alone adequately explains the disease phenotype.
Inflammatory Pathway
- CD8+ cytotoxic T lymphocytes (CTLs) invade non-necrotic muscle fibres, forming endomysial inflammatory infiltrates.
- Upregulation of MHC class I on muscle fibres (often referred to as the "MHC-I stress response") amplifies the immune recognition.
- Perforin and granzyme-mediated cytotoxicity contributes to muscle fibre damage.
- Macrophage and dendritic cell activation further sustains chronic inflammation.
- Despite this inflammatory infiltrate, conventional immunosuppression is ineffective — suggesting the inflammation may be secondary or self-perpetuating.
Degenerative / Protein Aggregation Pathway
- Rimmed vacuoles: autophagic vacuoles with basophilic granular material on H&E staining, containing myeloid debris and lysosomal markers.
- Protein misfolding and aggregation: deposition of amyloid-β (Aβ), phosphorylated tau, p62/SQSTM1, and TDP-43 within vacuolated muscle fibres — sharing pathological features with neurodegenerative conditions such as Alzheimer's disease and frontotemporal dementia.
- Endoplasmic reticulum (ER) stress: unfolded protein response activation contributes to fibre atrophy and dysfunction.
- Mitochondrial dysfunction: COX (cytochrome c oxidase)-negative ragged-red and ragged-blue fibres reflect age-related mitochondrial DNA mutations compounded by inflammatory oxidative stress.
- Myonuclear degeneration: nuclear membrane abnormalities and TDP-43 mislocalisation suggest intrinsic nuclear dysfunction.
Selective Pattern of Muscle Weakness
The clinical hallmark of IBM is the selective involvement of specific muscle groups, producing a pattern that is distinct from other inflammatory myopathies:
| Muscle Group Affected | Clinical Consequence | Frequency |
|---|---|---|
| Finger flexors (FDP) | Difficulty gripping, opening jars, turning keys, pinch weakness | ~80–95% |
| Quadriceps (knee extensors) | Difficulty rising from chairs, climbing stairs, frequent falls | ~80–95% |
| Wrist flexors | Wrist drop, difficulty carrying objects | ~50–70% |
| Ankle dorsiflexors (tibialis anterior) | Foot drop, steppage gait, trip hazard | ~50–70% |
| Pharyngeal / cricopharyngeal muscles | Dysphagia, aspiration risk, weight loss | 40–60% |
| Proximal upper limb (deltoid, biceps) | Difficulty lifting arms overhead | ~40–50% |
| Neck flexors | Difficulty lifting head from pillow | ~30–40% |
Key Clinical Features
- Asymmetric onset: weakness is often noticeably worse on one side, particularly in the upper limbs.
- Slow progression: the disease evolves over years; patients often recall subtle difficulty with buttons or mild knee buckling long before presentation.
- Dysphagia (40–60%): may be the presenting complaint. Caused by cricopharyngeal dysfunction and pharyngeal muscle weakness. Contributes to malnutrition, aspiration pneumonia, and reduced quality of life. Dysphagia may occur early or even precede limb symptoms in some cases.
- Preservation of some muscle groups: notably, shoulder abductors and hip flexors may be relatively spared early in the disease course.
- No skin involvement: unlike dermatomyositis, there are no rashes, heliotrope discolouration, or Gottron papules.
- Minimal pain: IBM is typically painless, distinguishing it from polymyalgia rheumatica and many musculoskeletal conditions.
- Systemic features absent: no fever, no interstitial lung disease (unlike anti-synthetase syndrome), no Raynaud's phenomenon.
- Association with other conditions: IBM is associated with autoimmune diseases (Sjögren syndrome, systemic lupus erythematosus, sarcoidosis), haematological malignancy, and HIV/HTLV-1 infection in some populations.
Diagnosis
Diagnostic Criteria
Multiple diagnostic criteria sets have been proposed. The 2011 European Neuromuscular Centre (ENMC) criteria and the 2017 ENMC revised criteria are the most widely used in Australian neuromuscular practice. Diagnosis requires a combination of clinical, laboratory, and pathological features.
Laboratory Investigations
Creatine Kinase (CK)
- CK is mildly elevated (typically 2–4× ULN) in the majority of patients.
- CK may be normal in 10–20% of cases — a normal CK does not exclude IBM.
- Markedly elevated CK (>10× ULN) should prompt consideration of an alternative diagnosis (e.g., immune-mediated necrotising myopathy).
Anti-cN-1A (NT5C1A) Antibodies
- Sensitivity 30–60% depending on assay and cohort.
- Specificity ~85–95% for IBM among inflammatory myopathies.
- A positive result supports the diagnosis; a negative result does not exclude IBM.
- Available through Australian referral laboratories (including hospital immunology and commercial reference labs).
- Higher titres may correlate with more severe dysphagia and more aggressive disease.
Other Blood Tests
- ESR and CRP are typically normal or mildly elevated.
- ANA, ENA, myositis-specific antibodies (MSAs) and myositis-associated antibodies (MAAs) — primarily to exclude other inflammatory myopathies.
- Consider HTLV-1 and HIV serology in appropriate clinical context.
Electrodiagnostic Studies
Muscle MRI
- Demonstrates fatty infiltration and oedema in characteristic patterns.
- Quadriceps: selective involvement of vastus medialis and vastus lateralis with relative sparing of rectus femoris.
- Forearm: selective fatty replacement of flexor digitorum profundus (FDP) — a highly suggestive finding.
- STIR signal (oedema) indicates active inflammation; T1 hyperintensity indicates chronic fatty replacement.
- MRI can guide biopsy site selection to maximise diagnostic yield.
Muscle Biopsy — Gold Standard
Biopsy of a moderately affected muscle (avoid end-stage fatty replacement) yields the highest diagnostic sensitivity. Histopathological features include:
- Endomysial inflammatory infiltrates: CD8+ T cells surrounding and invading non-necrotic muscle fibres.
- Rimmed vacuoles: basophilic granular material rimming sarcoplasmic vacuoles (seen on modified Gomori trichrome stain). Present in 60–90% of biopsies.
- COX-negative fibres: cytochrome c oxidase-deficient fibres reflecting mitochondrial dysfunction. Increased in frequency with age and disease duration.
- Ragged-red fibres: modified Gomori trichrome-positive fibres indicating subsarcolemmal mitochondrial accumulation.
- p62/SQSTM1 and TDP-43 inclusions: immunohistochemical staining reveals characteristic sarcoplasmic aggregates — relatively specific for IBM.
- Amyloid-β and phosphorylated tau deposits: Congo red positivity (under rhodamine optics) and tau immunostaining within vacuolated fibres.
- MHC class I upregulation: sarcolemmal expression of MHC-I in affected fibres.
- Fibre size variation: atrophic and hypertrophic fibres, often with fibre-type grouping suggesting chronicity.
Differential Diagnosis
| Condition | Key Distinguishing Features |
|---|---|
| Polymyositis | Symmetric proximal weakness; responds to steroids; no rimmed vacuoles |
| Dermatomyositis | Skin rash; perifascicular atrophy on biopsy; responds to immunosuppression |
| Anti-synthetase syndrome | Myositis + ILD + mechanic's hands + anti-Jo-1 |
| Immune-mediated necrotising myopathy | High CK; necrotising fibres without inflammation; anti-SRP or anti-HMGCR |
| Limb-girdle muscular dystrophy | Family history; genetic testing; no inflammation on biopsy |
| Motor neuron disease (ALS) | Fasciculations; upper motor neuron signs; denervation on EMG without myopathic units |
| Late-onset Pompe disease | Proximal weakness + respiratory failure; acid α-glucosidase assay; vacuolar glycogen storage |
| Polymyalgia rheumatica | Pain > weakness; elevated ESR/CRP; responds rapidly to low-dose prednisolone |
Management & Prognosis
Pharmacological Management
Current evidence does not support routine use of immunosuppressive therapy in IBM. However, some clinicians offer a time-limited trial (3–6 months) of therapy in cases where diagnostic uncertainty exists or the phenotype overlaps with treatable inflammatory myopathy. The following agents have been studied:
Emerging therapies under investigation: Several targeted therapies are in clinical trials, including anti-TGF-β (LY364947/bimagrumab-related), JAK inhibitors (tofacitinib, ruxolitinib), anti-CD40 antibodies, and sirolimus (mTOR inhibition to enhance autophagy). None are currently approved or PBS-listed for IBM. Patients may be referred for consideration of clinical trial enrolment at major Australian neuromuscular centres (e.g., Royal Prince Alfred, Austin Health, Royal Adelaide Hospital).
Non-Pharmacological Management — The Mainstay
Physiotherapy & Exercise
- Progressive resistance training: safe and may modestly slow functional decline. Should be supervised by a physiotherapist experienced in neuromuscular conditions.
- Aerobic exercise: stationary cycling, swimming, walking programmes maintain cardiovascular fitness and functional mobility.
- Falls prevention programmes: home modification assessment, balance training, and quadriceps strengthening are essential given the high fall rate.
- Stretching and range-of-motion exercises: prevent contractures, particularly of the knee flexors and ankle plantarflexors.
- Avoid overexertion: excessive eccentric exercise may worsen muscle damage; exercise should be graded and progressive.
Occupational Therapy
- Adaptive equipment for hand weakness: jar openers, key turners, built-up handles, button hooks.
- Home modifications: grab rails, shower chairs, raised toilet seats.
- Wheelchair assessment and prescription when ambulation becomes unsafe.
- Assistive technology for grip-dependent tasks (smartphone stylus, voice-activated devices).
Dysphagia Management
- Speech-language pathology assessment: clinical bedside swallow assessment and videofluoroscopic swallow study (VFSS) or fibreoptic endoscopic evaluation of swallowing (FEES).
- Diet modification: texture-modified diets (IDDSI framework), thickened fluids as indicated.
- Swallowing manoeuvres: chin-tuck, effortful swallow, supraglottic swallow techniques.
- Cricopharyngeal myotomy: may improve pharyngeal phase dysphagia in selected patients; evidence is limited to case series.
- PEG (percutaneous endoscopic gastrostomy) tube: consider when weight loss >10%, recurrent aspiration, or severe dysphagia limiting oral intake. PEG does not prevent aspiration of oral secretions but ensures adequate nutrition.
- IVIg for dysphagia: a small RCT (Dalakas et al., 2001) showed marginal improvement in swallowing function; some centres offer a therapeutic trial in patients with severe dysphagia despite its lack of effect on limb strength.
Orthotics & Mobility Aids
- Ankle-foot orthosis (AFO): for foot drop — reduces trip/fall risk. Custom-moulded AFOs preferred. Available under Australian state/territory orthotics programmes.
- Wrist splints: for wrist drop; improve hand function during activities.
- Walking aids: quad sticks, rollator frames, and eventually wheelchairs as disease progresses.
- Standing frames: for patients with severe quadriceps weakness who are transitioning from ambulatory to wheelchair status.
Monitoring & Follow-Up
Prognosis
- IBM is a slowly progressive disease with no spontaneous remissions.
- Walking aids: most patients require a walking aid (cane, frame) within 10–15 years of symptom onset.
- Wheelchair dependence: approximately 30% of patients are wheelchair-dependent at 10 years from diagnosis.
- Dysphagia progression: swallowing worsens over time; recurrent aspiration is the major threat to survival.
- Mortality: excess mortality is largely attributable to aspiration pneumonia, respiratory failure (from diaphragmatic involvement in severe cases), and complications of immobility. Median survival from diagnosis is approximately 10–16 years in most cohorts.
- Quality of life: significantly impaired, driven by loss of independence, dysphagia, falls, and reduced social participation. Psychological support and peer-group connections (e.g., Muscular Dystrophy Australia, Myositis Association) are important.
Special Populations
ATSI Health Considerations
📚 References
- 1. Greenberg SA. Inflammatory myopathies: disease mechanisms. Curr Opin Neurol. 2019;32(5):681–687.
- 2. Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044–1055.
- 3. Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology. 2014;83(5):426–433.
- 4. Benveniste O, Guiguet M, Freebody J, et al. Long-term observational study of sporadic inclusion body myositis. Brain. 2011;134(Pt 11):3176–3184.
- 5. Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K. Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurology. 1997;48(3):712–716.
- 6. Dalakas MC, Koffman B, Fujii M, Spector S, Sivakumar K, Cupler E. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology. 2001;56(3):323–327.
- 7. Needham M, Corbett A, Day T, Christiansen F, Fabian V, Mastaglia FL. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci. 2008;15(12):1350–1353.
- 8. Larsson HE, Lindberg C, Lindvall B. A 10-year follow-up of patients with inclusion body myositis. Acta Neurol Scand. 2020;142(3):264–271.
- 9. Alexanderson H, Lundberg IE. Exercise as a therapeutic modality in patients with idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2012;24(2):201–207.
- 10. Lilleker JB, Rastansky S, Dey M, et al. Anti-cN-1A autoantibodies in sporadic inclusion body myositis: prevalence, clinical associations and validation of a new diagnostic assay. Neuromuscul Disord. 2021;31(8):721–729.
- 11. Theadom A, Rodrigues M, Poke G, et al. A nationwide, population-based depiction of the burden of muscular dystrophies in New Zealand. Neuroepidemiology. 2019;52(1-2):9–16.
- 12. Schmidt K, Kleber S, Schoser B, et al. Anti-cN-1A antibody positive patients with IBM show more severe dysphagia and higher mortality. Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e741.
- 13. Alemo Munters L, Alexanderson H, Crofford LJ, Lundberg IE. New insights into the benefits of exercise for muscle health in patients with idiopathic inflammatory myopathies. Rheumatology (Oxford). 2014;53(7):1165–1174.
- 14. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2020 summary report. Canberra: AIHW; 2020.
- 15. Muscular Dystrophy Australia. Inclusion body myositis: information for patients and families. Melbourne: MDA; 2023. Available at: www.mda.org.au.