📋 Key Information Summary
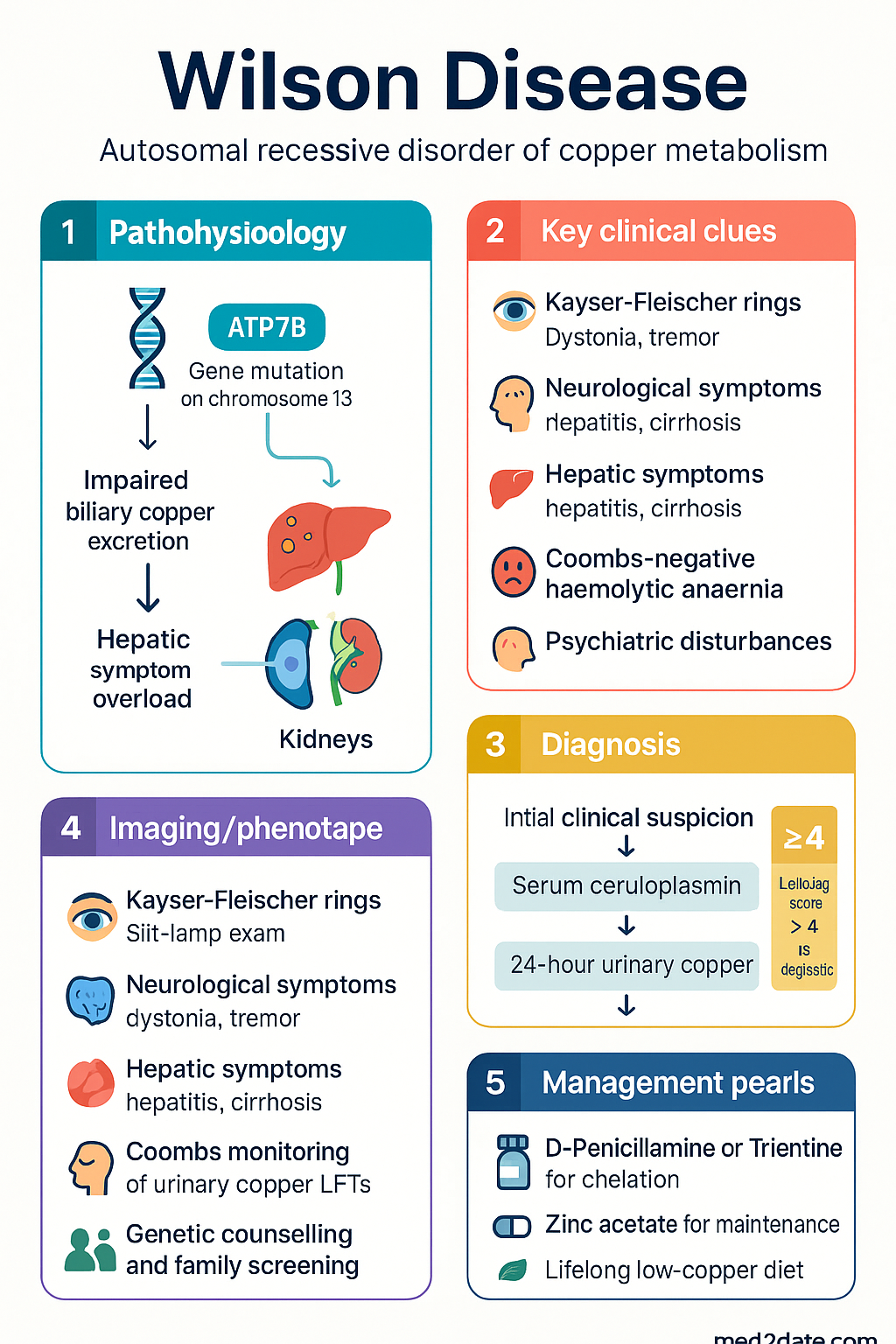
- Wilson disease is an autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene on chromosome 13, leading to impaired biliary copper excretion and toxic copper accumulation in liver, brain, cornea, and kidneys.
- Australian prevalence approximately 1 in 30,000; carrier frequency ~1 in 90. Higher consanguinity rates in some Aboriginal and Torres Strait Islander communities may increase local prevalence.
- Diagnosis uses the Leipzig (Ferenci) scoring system — a composite score incorporating ceruloplasmin, urinary copper, Kayser-Fleischer rings, liver copper, and ATP7B genotyping. Score ≥4 is diagnostic.
- Key diagnostic tests: serum ceruloplasmin <0.2 g/L, 24-hour urinary copper >1.6 µmol/24h (baseline), >25 µmol/24h after penicillamine challenge in children, and liver biopsy copper >250 µg/g dry weight.
- Kayser-Fleischer rings — golden-brown corneal deposits seen on slit-lamp examination — present in ~95% of neurological Wilson disease but only ~50% of hepatic presentations.
- First-line treatment: D-penicillamine (with concurrent pyridoxine 25 mg/day) 1–1.5 g/day in divided doses; risk of paradoxical neurological worsening in first weeks of therapy.
- Second-line chelator: trientine (triethylenetetramine dihydrochloride) 750–1500 mg/day, better tolerated with fewer adverse effects; pregnancy data more limited than penicillamine.
- Zinc acetate 150 mg elemental zinc/day (in 3 divided doses) as maintenance therapy or first-line in paucisymptomatic patients; acts by inducing intestinal metallothionein to block copper absorption.
- All patients require lifelong low-copper diet — avoid organ meats, shellfish, nuts (especially cashews), mushrooms, and chocolate.
- Wilsonian acute liver failure (ALF) is a medical emergency: typically young women, Coombs-negative haemolytic anaemia, ALP:bilirubin ratio <4, AST:ALT ratio >2.2, low serum uric acid, and very high mortality without urgent liver transplant.
- King's College Wilson Index predicts need for transplant: score ≥11 (or bilirubin >300 µmol/L with INR >6.5) mandates urgent transplant listing.
- Monitoring: 24-hour urinary copper (target 3–8 µmol/day on chelation), LFTs, FBC, direct Coombs test, and neurological assessment regularly. Non-compliance risk is lifelong.
- ATSI populations face diagnostic delays due to limited specialist and slit-lamp access in remote communities; culturally appropriate genetic counselling and family screening are essential.
Introduction & Australian Epidemiology
Wilson disease (hepatolenticular degeneration) is an autosomal recessive disorder of copper homeostasis caused by biallelic pathogenic variants in the ATP7B gene (chromosome 13q14.3). The encoded copper-transporting ATPase is expressed predominantly in hepatocytes and is essential for incorporating copper into caeruloplasmin and for biliary copper excretion. Loss of ATP7B function results in toxic hepatic copper accumulation, spillover into the systemic circulation, and deposition in the basal ganglia, cornea, kidneys, and other tissues.
Australian Epidemiology
- Estimated prevalence: 1 in 30,000 live births; carrier frequency approximately 1 in 90 across most Australian populations.
- The disease is pan-ethnic but certain founder mutations may be enriched in specific communities. Over 800 ATP7B variants are catalogued; H1069Q (European) and R778L (East Asian) are the most common worldwide.
- In Aboriginal and Torres Strait Islander communities, higher rates of consanguinity in some regions may increase homozygosity for recessive conditions including Wilson disease, though population-level prevalence data are limited.
- Presentation age is typically 5–35 years (hepatic form earlier, neurological form later), but cases diagnosed in the sixth decade are well documented.
- The AIHW National Notifiable Diseases Surveillance System does not capture Wilson disease; true Australian incidence is inferred from tertiary centre registries and transplant data.
- Australian liver transplant registries report Wilson disease as an indication in approximately 3–5% of paediatric transplants and 1–2% of adult transplants annually.
Pathophysiology
Copper is absorbed in the proximal small intestine and transported to the liver bound to albumin and transcuprein. In healthy hepatocytes, ATP7B shuttles copper to the trans-Golgi network for incorporation into apocaeruloplasmin and, when copper is replete, trafficks to the canalicular membrane for biliary excretion (the major route of copper elimination). In Wilson disease, ATP7B dysfunction causes:
- Failure to secrete copper into bile → hepatic copper overload → oxidative damage, steatosis, hepatitis, fibrosis, cirrhosis.
- Failure to incorporate copper into apocaeruloplasmin → accelerated degradation of apocaeruloplasmin → low serum caeruloplasmin.
- Non-caeruloplasmin-bound copper spilling into plasma → deposition in brain (lenticular nucleus, putamen, caudate), cornea (Descemet membrane → Kayser-Fleischer rings), kidneys (proximal tubular damage → aminoaciduria, Fanconi-like syndrome), and red blood cells (acute haemolysis).
- Cerebral copper toxicity produces the characteristic neuropsychiatric manifestations: dystonia, dysarthria, tremor, psychiatric disturbance (depression, psychosis, behavioural change).
Diagnosis
No single test is pathognomonic for Wilson disease. Diagnosis rests on a combination of clinical, biochemical, histological, and genetic findings, scored using the Leipzig (Ferenci) Scoring System. A cumulative score of ≥4 is considered diagnostic.
Leipzig (Ferenci) Scoring System
| Parameter | Finding | Score |
|---|---|---|
| Kayser-Fleischer rings | Present on slit-lamp (experienced observer) | 2 |
| Absent | 0 | |
| Present but examination not available | 1 | |
| Serum caeruloplasmin | <0.1 g/L | 2 |
| 0.1–0.2 g/L | 1 | |
| >0.2 g/L | 0 | |
| Coombs-negative haemolytic anaemia | Present | 1 |
| Absent | 0 | |
| 24-hour urinary copper (baseline) | Above normal (>1.6 µmol/24h or >100 µg/24h) | 1 |
| Normal but >1× ULN after D-penicillamine challenge* | 1 | |
| Normal | 0 | |
| Liver copper quantification (biopsy) | >250 µg/g dry weight (5 µmol/g) | 2 |
| 50–250 µg/g dry weight | 1 | |
| Normal (<50 µg/g dry weight) — stain negative | −1 | |
| Rhodamine-positive hepatocytes (histochemistry) | Positive | 1 |
| Negative | 0 | |
| ATP7B mutation analysis | Homozygous or compound heterozygous mutation | 4 |
| Heterozygous mutation (1 allele detected) | 1 | |
| No mutation detected | 0 |
Key Diagnostic Investigations
Diagnostic Approach by Clinical Presentation
- Chronic hepatitis mimicking autoimmune hepatitis
- Steatohepatitis (non-alcoholic pattern)
- Cryptogenic cirrhosis
- Fulminant hepatic failure with haemolysis (see Acute Liver Failure section)
- Kayser-Fleischer rings may be absent (~50%)
- Resting or wing-beating tremor
- Dystonia (risus sardonicus, dysarthria)
- Dysphagia, sialorrhoea
- Psychiatric: depression, anxiety, psychosis, behavioural change
- Kayser-Fleischer rings usually present (~95%)
Treatment
Treatment is lifelong and must be initiated as soon as the diagnosis is confirmed, regardless of symptoms. The goals of therapy are to remove accumulated copper, prevent further copper deposition, and manage symptoms. All patients require dietary modification, and most require pharmacological copper chelation or zinc therapy.
Copper Chelators
Zinc Therapy
Treatment Strategy
Low-Copper Diet
Dietary copper restriction is an essential adjunct to pharmacotherapy in all Wilson disease patients.
- Organ meats (liver, kidney) — very high copper
- Shellfish (oysters, crab, lobster, mussels)
- Nuts — especially cashews, Brazil nuts, sunflower seeds
- Chocolate and cocoa
- Mushrooms (dried especially)
- Soy products (tofu, soy milk)
- Dried fruits (prunes, figs)
- Multivitamins with copper
- Copper cookware; water from copper pipes (first morning flush)
- Meat (muscle, not organ) — moderate copper
- Dairy products
- Eggs
- White bread, rice, pasta
- Most fruits and vegetables (except those listed to avoid)
- Tea and coffee (in moderation)
Monitoring Protocol
| Test | Frequency | Target / Interpretation |
|---|---|---|
| 24-hour urinary copper | Monthly × 6 months, then 3-monthly, then annually | On chelator: 3–8 µmol/day (200–500 µg/day). <2 µmol/day on chelator = non-compliance risk. On zinc: 0.5–1.5 µmol/day. |
| LFTs (ALT, AST, GGT, ALP, bilirubin, albumin) | Monthly initially → 3-monthly → annual | Normalisation within 6–12 months expected. Rising ALT may indicate non-compliance or inadequate therapy. |
| FBC, reticulocyte count | Monthly × 6 months (penicillamine), then 3-monthly | Penicillamine: watch for thrombocytopaenia, leukopaenia, sideroblastic anaemia (trientine). |
| Serum free copper | 3-monthly initially → annual | Target <1.6 µmol/L. Rising values suggest non-compliance or inadequate dose. |
| Urinalysis + urine protein | Monthly (penicillamine) → 3-monthly | Penicillamine nephrotoxicity: proteinuria >1 g/day warrants dose review. |
| Neurological examination | 3-monthly initially → 6-monthly | Objective assessment of tremor, dystonia, dysarthria. Document baseline before treatment. |
| Slit-lamp examination | Annual | KF rings may fade with effective therapy. Persistence suggests inadequate decoppering. |
Acute Liver Failure
Wilsonian acute liver failure (ALF) accounts for approximately 5–12% of all non-paracetamol ALF cases in Australian transplant centres. It represents the most immediately life-threatening presentation of Wilson disease and requires rapid recognition and urgent transplant assessment.
Clinical Features of Wilsonian ALF
Wilsonian ALF has a distinctive biochemical and clinical profile that differs from other causes of ALF:
- Demographics: Typically young women (female: male ratio ~2:1), age 12–30 years (though can occur at any age).
- Coombs-negative haemolytic anaemia: Present in virtually all cases. Acute copper release from necrotic hepatocytes causes direct oxidative haemolysis. Reticulocytosis may be present. Direct Coombs (DAT) is negative.
- Very high bilirubin with disproportionately low ALP: The hallmark ratio — ALP (IU/L) / total bilirubin (mg/dL) <4 [or ALP:bilirubin (µmol/L) ratio <0.57]. This reflects the combination of haemolysis-driven hyperbilirubinaemia and falling hepatic synthetic production of ALP.
- AST > ALT: AST:ALT ratio >2.2 is characteristic. Both transaminases are typically elevated but rarely exceed 2000 IU/L (unlike paracetamol ALF).
- Low serum uric acid: Copper-induced proximal tubular dysfunction causes renal uric acid wasting. Hypouricaemia is a useful diagnostic clue.
- Low caeruloplasmin: Usually very low, but can be misleadingly normal due to acute-phase response.
- Kayser-Fleischer rings: Usually present in ALF (often absent in chronic hepatic presentations, but the acute decompensation usually occurs in patients who already have established copper deposition).
- Rapidly progressive: Jaundice → coagulopathy → hepatic encephalopathy → multi-organ failure within days to weeks.
Diagnostic Clue: The ALP:Bilirubin Ratio
King's College Wilson Index (Modified)
The King's College Criteria for Wilsonian ALF use a scoring system to predict the need for urgent liver transplantation. The score is calculated at presentation:
| Parameter | Score |
|---|---|
| Serum bilirubin (µmol/L) | Bilirubin / 100 (= points) |
| AST (IU/L) | AST / 100 (= points) |
| INR | INR × 100 (= points) |
| WBC (×10⁹/L) | WBC × 10 (= points) |
| ALP:bilirubin ratio (conventional units) | Ratio <4 = additional predictor |
Immediate Management of Wilsonian ALF
Differential Diagnosis of ALF with Haemolysis
| Condition | Key Distinguishing Feature |
|---|---|
| Wilsonian ALF | Coombs-negative haemolysis, ALP:bilirubin <4, low uric acid, KF rings, AST:ALT >2.2 |
| Autoimmune hepatitis flare | Positive autoantibodies (ANA, ASMA, anti-LKM-1), elevated IgG, Coombs may be positive |
| Haemophagocytic lymphohistiocytosis | Very high ferritin, cytopenias, hypertriglyceridaemia, low fibrinogen, haemophagocytes on biopsy |
| Paracetamol toxicity | Very high AST/ALT (>3000), paracetamol level, history, no haemolysis |
| Budd-Chiari syndrome | Doppler US showing hepatic vein thrombosis, hepatomegaly, ascites |
| Pregnancy-related ALF (AFLP, HELLP) | Pregnancy setting, HELLP = haemolysis, elevated LFTs, low platelets; delivery is treatment |
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Wilson disease affects Aboriginal and Torres Strait Islander peoples. While population-level prevalence data are limited, the clinical impact is amplified by diagnostic delays, barriers to specialist access, and the need for lifelong treatment adherence in communities with competing health priorities.
📚 References
- 1. European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. 2012;56(3):671–685. doi:10.1016/j.jhep.2011.11.007
- 2. European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406–460. doi:10.1016/j.jhep.2018.03.024
- 3. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139–142. doi:10.1034/j.1600-0676.2003.00824.x
- 4. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089–2111. doi:10.1002/hep.22261
- 5. Schilsky ML. Wilson disease: diagnosis, treatment, and follow-up. Clin Liver Dis. 2017;21(4):755–767. doi:10.1016/j.cld.2017.06.011
- 6. Dhawan A, Taylor RM, Cheeseman P, et al. Wilson's disease in children: 37-year experience and revised King's College scores for liver transplantation. Liver Transpl. 2005;11(4):441–448. doi:10.1002/lt.20352
- 7. Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers. 2018;4(1):21. doi:10.1038/s41572-018-0018-3
- 8. Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369(9559):397–408. doi:10.1016/S0140-6736(07)60196-2
- 9. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework. Canberra: AIHW; 2023. Available from: https://www.aihw.gov.au
- 10. Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice (Red Book). 10th edn. East Melbourne: RACGP; 2023.
- 11. Bruha R, Marecek Z, Pospisilova L, et al. Long-term follow-up of Wilson disease patients: a 25-year experience. QJM. 2021;114(12):851–857. doi:10.1093/qjmed/hcab086
- 12. Weiss KH, Thurik F, Gotthardt DN, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11(8):1028–1035. doi:10.1016/j.cgh.2013.03.017
- 13. Brewer GJ, Dick RD, Johnson VD, et al. Treatment of Wilson's disease with zinc: XV long-term follow-up studies. J Lab Clin Med. 1998;132(4):264–278. doi:10.1016/S0022-2143(98)90039-7
- 14. Australian Commission on Safety and Quality in Health Care (ACSQHC). National Safety and Quality Health Service Standards. 2nd edn. Sydney: ACSQHC; 2021.
- 15. RHDAustralia. Recommendations for Aboriginal and Torres Strait Islander peoples. Darwin: RHDAustralia; 2022. Available from: https://www.rhdaustralia.org.au