📋 Key Information Summary
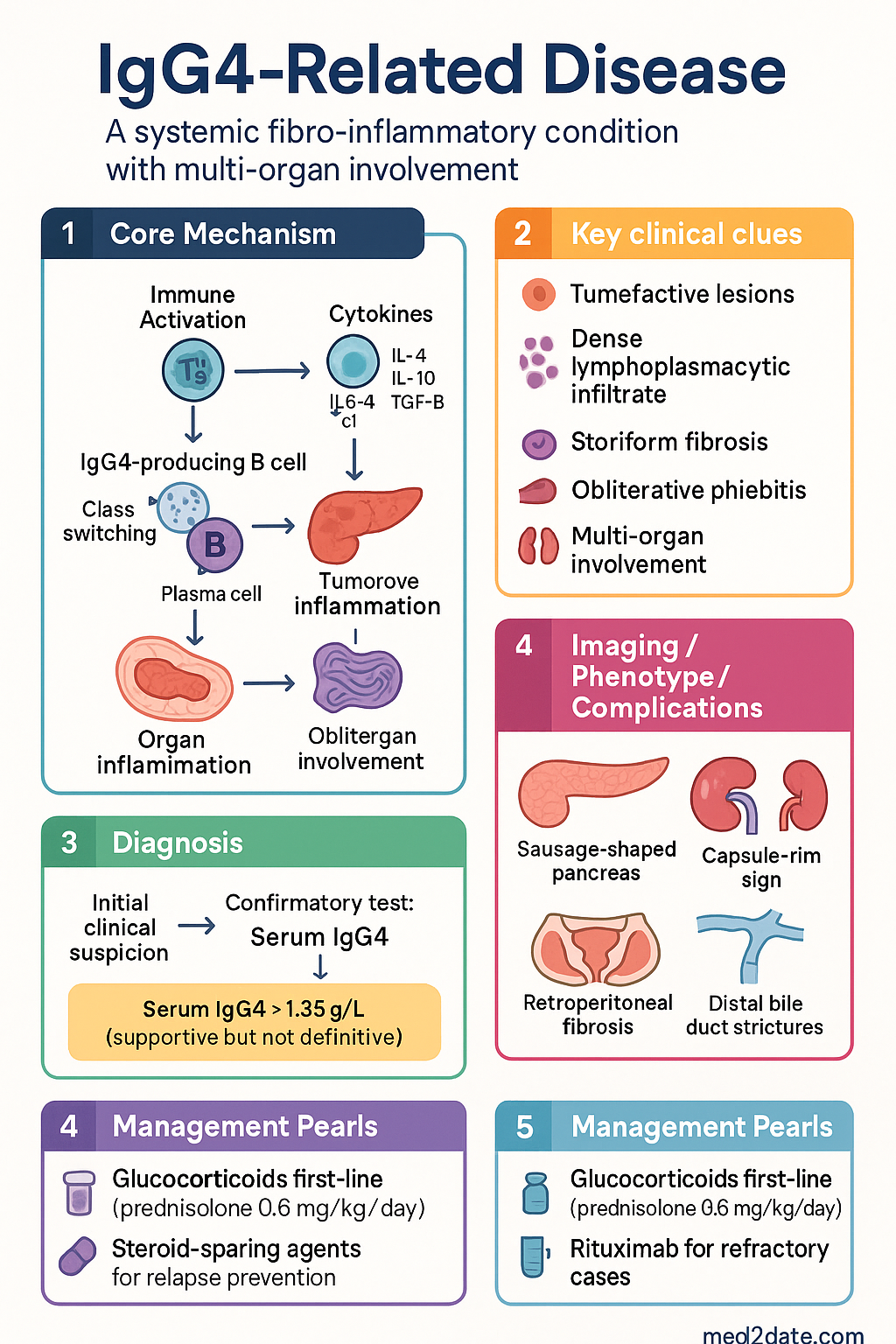
- IgG4-related disease (IgG4-RD) is a systemic fibro-inflammatory condition characterised by tumefactive lesions, a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform fibrosis, and obliterative phlebitis.
- Diagnosis requires clinicopathological correlation: serum IgG4 >1.35 g/L is supportive but can be normal in up to 30–40% of confirmed cases; histopathology remains the gold standard.
- Histopathological triad: lymphoplasmacytic infiltrate, storiform (cartwheel) fibrosis, and obliterative phlebitis with an IgG4+/IgG+ plasma cell ratio >40% on immunostaining.
- ACR/EULAR 2019 classification criteria require entry criterion, exclusion criteria, and weighted domains (clinical, serological, imaging, histopathological) yielding a score ≥20 for classification.
- Type 1 autoimmune pancreatitis (AIP) is the most common pancreatic manifestation; imaging shows a diffusely enlarged "sausage-shaped" pancreas with peripancreatic capsule-rim sign.
- IgG4-related sclerosing cholangitis mimics primary sclerosing cholangitis and cholangiocarcinoma; long-segment distal bile duct strictures with associated AIP favour IgG4-RD.
- Glucocorticoids are first-line therapy: prednisolone 0.6 mg/kg/day induction (typically 30–40 mg), tapered over 3–6 months with planned maintenance for 3 years in Japan/Australia.
- Steroid-sparing agents (azathioprine, mycophenolate mofetil, methotrexate) are used for relapse prevention or when glucocorticoid toxicity is a concern.
- Rituximab (anti-CD20, 1000 mg IV ×2 doses fortnightly) is highly effective for refractory or relapsing disease and emerging as first-line in some protocols.
- Multi-organ involvement is the rule: pancreas, biliary tree, salivary/lacrimal glands, retroperitoneum, kidneys, aorta, lungs, and orbits may all be affected simultaneously or metachronously.
- Monitor treatment response with serial serum IgG4 levels (but note IgG4 may remain elevated despite clinical remission), repeat imaging at 4–8 weeks, and CRP/ESR.
- Malignancy must be excluded before initiating immunosuppression; pancreatic cancer, cholangiocarcinoma, and lymphoma are key differential diagnoses.
- Relapse rates are 30–50% after glucocorticoid cessation; maintenance immunosuppression or rituximab reduces recurrence.
- Aboriginal and Torres Strait Islander peoples may have delayed diagnosis due to healthcare access barriers; increased vigilance is required in communities with high autoimmune disease prevalence.
🎧 Audio Brief
Introduction & Australian Epidemiology
IgG4-related disease (IgG4-RD) is a recently recognised immune-mediated fibro-inflammatory condition that can affect nearly every organ system. Originally described in the context of autoimmune pancreatitis, it is now understood as a distinct multisystem entity unified by characteristic histopathological features and elevated tissue IgG4-positive plasma cells.
The disease predominantly affects middle-aged to elderly men (male-to-female ratio approximately 2–3:1), with peak incidence in the 6th–7th decade of life. In Australia, the estimated prevalence is 5–10 per 100,000 population, though this is likely an underestimate given diagnostic challenges and under-recognition.
Key epidemiological points relevant to Australian practice:
- Type 1 autoimmune pancreatitis accounts for the majority of IgG4-RD presentations to Australian gastroenterology and hepatopancreatobiliary (HPB) units.
- Retroperitoneal fibrosis is increasingly recognised as an IgG4-RD manifestation; the Prince Charles Hospital and Austin Health retroperitoneal fibrosis registries document growing case numbers.
- IgG4-RD may mimic malignancy in up to 50% of cases at initial presentation, leading to unnecessary surgical resections if tissue diagnosis is not pursued.
- The Australian national pathology laboratories (including Sonic Healthcare, Laverty, and public hospital pathology) have standardised serum IgG4 immunoassay availability since 2015.
Diagnosis
IgG4-RD diagnosis requires integration of clinical features, serological markers, imaging findings, and histopathology. No single test is pathognomonic; a multidisciplinary approach involving rheumatology, gastroenterology, pathology, and radiology is essential.
Serum IgG4
- Upper limit of normal: 1.35 g/L (may vary slightly between Australian laboratories; confirm local reference ranges).
- Sensitivity: approximately 60–70%; therefore 30–40% of patients with confirmed IgG4-RD have normal serum IgG4.
- Specificity: elevated IgG4 is not unique to IgG4-RD; levels may be raised in allergic conditions, infections, malignancy, and other autoimmune diseases.
- Elevated IgG4 >2× ULN (i.e., >2.7 g/L) has higher specificity for IgG4-RD.
- Serum IgG subclass analysis is available on request through major Australian referral laboratories (MBS item applicable).
Histopathology
Tissue diagnosis remains the gold standard. The characteristic histopathological triad includes:
| Feature | Description | Diagnostic Value |
|---|---|---|
| Lymphoplasmacytic infiltrate | Dense infiltrate of mature lymphocytes and plasma cells, often with lymphoid follicle formation | High sensitivity; present in virtually all cases |
| Storiform fibrosis | Spindle-cell proliferation in a "cartwheel" or swirling pattern | Highly characteristic; distinguishes from other inflammatory conditions |
| Obliterative phlebitis | Inflammation and fibrosis obliterating venous lumina; arteries are typically spared | Most specific feature; identified in 50–80% of biopsies |
Immunohistochemistry for IgG4-positive plasma cells:
- Absolute count: ≥10 IgG4-positive plasma cells per high-power field (HPF) in most organs; ≥50/HPF for pancreatic tissue.
- IgG4+/IgG+ ratio: >40% is strongly supportive of IgG4-RD.
- Core needle biopsy or surgical excision biopsy is preferred; fine-needle aspiration is generally insufficient for histopathological assessment.
ACR/EULAR 2019 Classification Criteria
The 2019 ACR/EULAR classification criteria for IgG4-RD provide a standardised framework for research and clinical classification:
- Entry criterion: At least one characteristic organ swelling/mass or organ dysfunction suggestive of IgG4-RD.
- Exclusion criteria: 32 items excluding mimicking conditions (e.g., malignancy, infections, other autoimmune diseases).
- Weighted inclusion criteria across four domains:
| Domain | Item | Score |
|---|---|---|
| Clinical | Symmetric lacrimal, parotid, or submandibular gland enlargement | +3 |
| Pancreas involvement (diffuse enlargement/rim/hypoenhancement) | +4 | |
| Biliary tract involvement | +3 | |
| Serological | IgG4 > ULN but ≤2× ULN | +4 |
| IgG4 > 2× ULN | +6 | |
| Imaging | Multiorgan involvement on imaging | +2 to +5 |
| Histopathological | Lymphoplasmacytic infiltrate + storiform fibrosis | +7 |
| IgG4+/IgG+ >40% and ≥10 IgG4+ cells/HPF | +6 |
Classification threshold: Total score ≥20 after exclusion criteria are met.
HISORt Criteria for Autoimmune Pancreatitis
The HISORt criteria (Histology, Imaging, Serology, Other organ involvement, Response to therapy) remain widely used in Australian HPB centres for Type 1 AIP diagnosis:
- H — Histology: Lymphoplasmacytic sclerosing pancreatitis on core biopsy or resection specimen.
- I — Imaging: Characteristic pancreas imaging (diffuse enlargement, delayed enhancement, capsule rim, or focal mass).
- S — Serology: Elevated serum IgG4 (>1.35 g/L) or other IgG subclass abnormalities.
- O — Other organ involvement: Biliary strictures, salivary gland enlargement, retroperitoneal fibrosis, renal lesions, etc.
- Rt — Response to therapy: Resolution or marked improvement with glucocorticoid trial (supports but does not establish diagnosis).
Diagnosis is established if any one group of criteria is fulfilled; combination of multiple groups strengthens diagnostic confidence.
GI & Hepatopancreatobiliary Manifestations
The gastrointestinal tract and hepatopancreatobiliary (HPB) system are the most commonly affected domains in IgG4-RD, accounting for approximately 60–70% of presentations in Australian tertiary centres.
Type 1 Autoimmune Pancreatitis (AIP)
Type 1 AIP is the pancreatic manifestation of IgG4-RD and the most common organ-specific presentation. It must be distinguished from Type 2 AIP (idiopathic duct-centric pancreatitis), which is not associated with IgG4 elevation or systemic involvement.
| Feature | Type 1 AIP (IgG4-related) | Type 2 AIP |
|---|---|---|
| Age | 60–70 years | 40–50 years |
| Sex ratio | M:F = 3:1 | M:F = 1:1 |
| Serum IgG4 | Elevated in 60–70% | Normal |
| Systemic involvement | Common (biliary, salivary, retroperitoneal) | Absent |
| Relapse rate | 30–50% | <10% |
| IBD association | Rare | Up to 30% |
Characteristic imaging findings (CT/MRI):
- Diffuse enlargement: "Sausage-shaped" pancreas with loss of normal lobular contour.
- Capsule-rim sign: Hypoenhancing rim of tissue surrounding the pancreas on portal venous phase CT; represents peripancreatic fibrosis/inflammation.
- Delayed enhancement: On dynamic CT or MRI, the pancreatic parenchyma shows delayed enhancement relative to normal pancreas.
- Focal mass form: May mimic pancreatic adenocarcinoma; absence of upstream duct dilatation, vascular encasement, or distant metastases favours AIP.
- MRCP: Long-segment distal CBD stricture with upstream biliary dilatation; pancreatic duct may be attenuated or irregular.
IgG4-Related Sclerosing Cholangitis (IgG4-SC)
IgG4-SC is the biliary manifestation of IgG4-RD and is present in approximately 60–90% of patients with Type 1 AIP. It is critical to distinguish IgG4-SC from primary sclerosing cholangitis (PSC) and cholangiocarcinoma, as treatment approaches differ fundamentally.
| Feature | IgG4-SC | PSC | Cholangiocarcinoma |
|---|---|---|---|
| Age at diagnosis | 60–70 years | 30–40 years | >60 years |
| Sex | Male predominance | Male predominance | Male predominance |
| Stricture pattern | Long-segment distal CBD; proximal intrahepatic uncommon | Multifocal, beaded intrahepatic | Focal, irregular, asymmetric |
| IgG4 level | Elevated | Usually normal | Occasionally elevated |
| Concurrent AIP | Present in ~80% | Absent | Absent |
| Response to steroids | Excellent | Minimal | None |
| Malignancy risk | Low | Cholangiocarcinoma risk 10–15% | IS the malignancy |
Retroperitoneal Fibrosis
- IgG4-RD accounts for approximately 50–60% of idiopathic retroperitoneal fibrosis cases.
- Presents as a peri-aortic soft-tissue mass (typically below the renal arteries) encasing the ureters and causing obstructive uropathy.
- MRI is preferred over CT for monitoring soft-tissue response; FDG-PET/CT may be used to assess disease activity.
- Bilateral ureteric stenting or nephrostomy may be necessary for acute obstructive renal failure prior to immunosuppression.
Sclerosing Sialadenitis & Other Head/Neck Involvement
- Küttner tumour: Chronic sclerosing sialadenitis of the submandibular gland presenting as a firm, painless mass mimicking malignancy.
- Mikulicz disease: Bilateral symmetrical enlargement of lacrimal, parotid, and submandibular glands — now recognised as IgG4-RD (distinct from Sjögren syndrome).
- Orbital involvement: Proptosis, restrictive strabismus, infraorbital nerve enlargement.
- Tissue biopsy is essential to exclude lymphoma and Sjögren syndrome.
Multi-Organ Involvement
IgG4-RD is inherently a multi-organ disease. In Australian series, approximately 40–60% of patients have involvement of two or more organs at diagnosis. Recognised sites include:
| Organ System | Manifestation | Key Investigation |
|---|---|---|
| Pancreas | Type 1 AIP | CT/MRI, EUS-FNB |
| Biliary tree | IgG4-SC | MRCP, ERCP with brushings |
| Retroperitoneum | Retroperitoneal fibrosis | CT/MRI abdomen-pelvis |
| Salivary/lacrimal glands | Sclerosing sialadenitis, Mikulicz | Ultrasound, biopsy |
| Kidneys | Tubulointerstitial nephritis, mass lesions | CT, renal biopsy |
| Aorta/periaortic tissue | Inflammatory aortic aneurysm, periaortitis | CT angiography, FDG-PET |
| Lungs | Nodules, bronchovascular thickening, pleural thickening | HRCT, VATS biopsy |
| Orbits | Orbital pseudotumour, dacryoadenitis | MRI orbits, biopsy |
| Meninges | Hypertrophic pachymeningitis | MRI brain with contrast |
| Thyroid | Riedel thyroiditis | Thyroid biopsy |
Investigations
A systematic investigative approach is essential for diagnosis, exclusion of mimics, and baseline assessment prior to treatment.
Treatment
Treatment of IgG4-RD aims to achieve remission of active inflammation, prevent organ damage from fibrosis, and manage relapses. Most patients respond dramatically to glucocorticoids, but relapse rates are significant and long-term immunosuppression strategies are often necessary.
First-Line: Glucocorticoid Induction
Expected response: Most patients show symptomatic and serological improvement within 2–4 weeks. Imaging response (reduction in organ swelling, resolution of capsule rim) is seen at 4–8 weeks. Prednisolone should be continued at the initial dose for 2–4 weeks before commencing taper.
Maintenance strategy (Australian approach): Following successful induction, many Australian centres maintain low-dose prednisolone (2.5–5 mg/day) for 1–3 years, particularly in patients with multi-organ involvement or elevated IgG4 at baseline. The Japanese consensus recommends 3-year maintenance; Australian practice varies.
Steroid-Sparing Agents
Steroid-sparing agents are indicated for: (1) relapse prevention, (2) steroid-dependent disease, (3) steroid intolerance/contraindications, and (4) as adjuncts to rituximab in refractory disease.
Second-Line / Refractory Disease: Rituximab
Rituximab (anti-CD20 monoclonal antibody) is increasingly used in IgG4-RD for relapsing, refractory, or steroid-intolerant disease. Depletion of B-cells addresses the central immunopathogenic mechanism and produces rapid, often dramatic, responses.
Treatment Algorithm
Monitoring Response
- Clinical: Symptom resolution (jaundice, gland swelling, mass-related symptoms) within 2–4 weeks of glucocorticoid initiation.
- Serological: Serum IgG4 typically falls within 4–8 weeks. However, IgG4 may remain elevated despite clinical remission in 30–50% of patients (so-called "serological non-responders"). Do not escalate therapy based on IgG4 alone if the patient is clinically well.
- Imaging: Repeat CT or MRI at 4–8 weeks post-induction. Reduction in organ swelling, resolution of capsule-rim sign, and improvement in biliary strictures are expected. Complete normalisation may take months.
- Biomarkers: CRP, ESR, and liver function tests (in IgG4-SC) provide complementary monitoring data.
- Relapse definition: Recurrence of symptoms, new organ involvement, or reappearance of imaging abnormalities after initial response. Fluctuation in IgG4 alone is not diagnostic of relapse.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Wallace ZS, Naden RP, Chari S, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis. 2020;79(1):77–87.
- 2. Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4(8):1010–1016.
- 3. Okazaki K, Kawa S, Kamisawa T, et al. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2013-I. Concept and diagnosis of autoimmune pancreatitis. J Gastroenterol. 2014;49(4):567–588.
- 4. Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67(7):1688–1699.
- 5. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385(9976):1460–1471.
- 6. Lanzillotta M, Mancuso G, Della-Torre E. Advances in the diagnosis and management of IgG4-related disease. BMJ. 2020;369:m1071.
- 7. Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171–1177.
- 8. Detlefsen S, Klöppel G. IgG4-related disease: with emphasis on the histopathological diagnosis. Stem Cell Investig. 2017;4:88.
- 9. Matsubayashi H, Uesaka K, Sugiura T, et al. IgG4-related sclerosing cholangitis and autoimmune pancreatitis. Hepatobiliary Surg Nutr. 2020;9(5):616–630.
- 10. Stone JH, Brito-Zerón P, Bosch X, Ramos-Casals M. Diagnostic approach to the complexity of IgG4-related disease. Mayo Clin Proc. 2015;90(7):927–939.
- 11. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework. Canberra: AIHW; 2023.
- 12. Australian Commission on Safety and Quality in Health Care (ACSQHC). National Safety and Quality Health Service Standards. 2nd ed. Sydney: ACSQHC; 2021.
- 13. Wallace ZS, Zhang Y, Perugino CA, et al. Clinical phenotypes of IgG4-related disease: an analysis of two international cross-sectional cohorts. Ann Rheum Dis. 2019;78(3):406–412.
- 14. Culver EL, Sadler R, Simpson D, et al. Elevated serum IgG4 levels in diagnosis, treatment response, organ involvement, and relapse in a prospective IgG4-related disease UK cohort. Am J Gastroenterol. 2017;112(4):733–743.
- 15. Royal Australasian College of Physicians (RACP). Clinical guidelines for the management of autoimmune conditions in Australia. Sydney: RACP; 2022.