📋 Key Information Summary
- Hereditary haemochromatosis (HH) is the most common inherited iron-overload disorder in Australia, predominantly affecting individuals of Northern European descent.
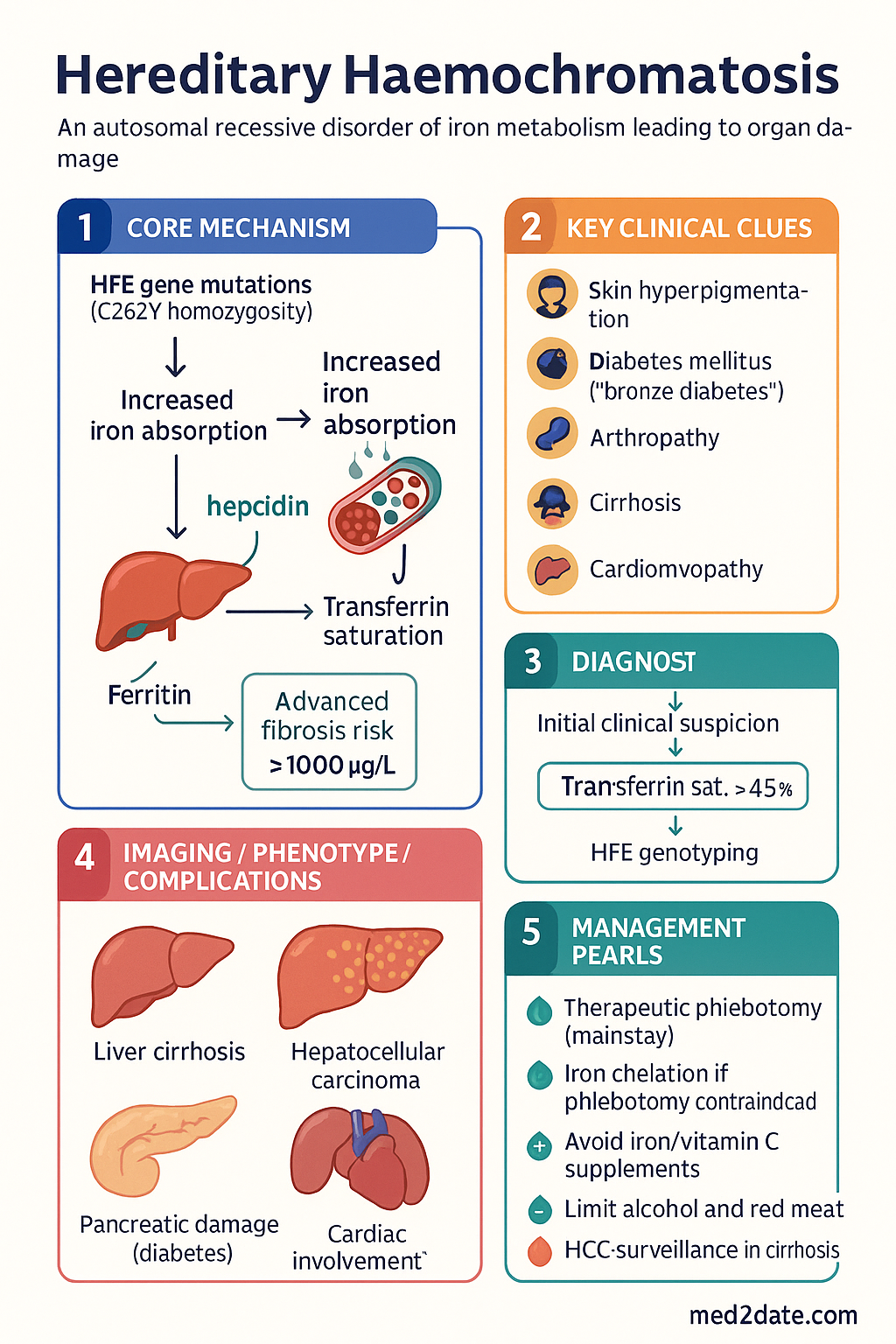
- C282Y homozygosity accounts for ≥90% of clinical HH cases; C282Y/H63D compound heterozygotes have lower penetrance and variable iron loading.
- Non-HFE forms (juvenile haemochromatosis with HJV/HAMP mutations, TFR2, ferroportin disease) are rare but cause more aggressive iron overload, often presenting in the 2nd–3rd decade.
- Diagnostic workup: Transferrin saturation ≥45% is the earliest biochemical marker; serum ferritin elevation confirms iron overload; HFE genotyping is the first-line confirmatory test.
- MRI T2* is the preferred non-invasive method to quantify hepatic iron concentration; liver biopsy is reserved for cases with ferritin >1000 µg/L with abnormal LFTs or suspected advanced fibrosis.
- Therapeutic phlebotomy is the cornerstone of treatment: weekly 450–500 mL venesections until serum ferritin reaches 50–100 µg/L, then maintenance every 2–4 months lifelong.
- Iron chelation with deferasirox (Jadenu®) is indicated when phlebotomy is contraindicated (severe anaemia, heart failure, poor venous access).
- Dietary advice: avoid iron and vitamin C supplements, minimise red meat alcohol excess, and avoid raw shellfish due to Vibrio vulnificus sepsis risk.
- First-degree relative screening (parents, siblings, children) with HFE genotyping and iron studies is mandatory once an index case is identified.
- Major complications include cirrhosis, hepatocellular carcinoma (HCC), diabetes mellitus, cardiomyopathy, hypogonadism, arthropathy, and skin hyperpigmentation.
- HCC surveillance (6-monthly AFP + ultrasound) is recommended for all patients with established cirrhosis or persistently elevated ferritin >1000 µg/L.
- Arthropathy preferentially affects the 2nd and 3rd metacarpophalangeal joints (MCPJs) with characteristic hooked osteophytes — a clinically distinctive finding.
- Aboriginal and Torres Strait Islander Australians should be screened where clinically indicated, though HH prevalence is lower than in non-Indigenous Australians of European descent.
- Early detection and pre-symptomatic treatment with phlebotomy can prevent all major complications and restore normal life expectancy.
Introduction & Australian Epidemiology
Hereditary haemochromatosis (HH) is an autosomal recessive disorder of iron metabolism characterised by progressive parenchymal iron deposition leading to organ damage. Australia has one of the highest prevalence rates of HFE-related haemochromatosis globally, owing to the Celtic and Northern European ancestry of much of the population.
In Australia, approximately 1 in 200 individuals of European descent are C282Y homozygous, and roughly 1 in 8 are heterozygous carriers. However, clinical penetrance is variable — not all homozygotes develop significant iron overload or organ damage. Males are typically affected earlier and more severely than females, with clinical manifestations usually appearing between ages 40 and 60 in men and post-menopause in women.
HH was historically called "bronze diabetes" due to the combination of skin hyperpigmentation and secondary diabetes mellitus. With earlier detection through population screening and family studies, most cases are now identified before irreversible organ damage occurs.
Genetics & Diagnosis
HFE Gene Mutations
The HFE gene on chromosome 6p21.3 encodes a protein that modulates hepcidin expression and thus controls systemic iron homeostasis. Two missense mutations account for the vast majority of clinical HH:
| Genotype | Prevalence (European) | Iron Overload Risk | Penetrance |
|---|---|---|---|
| C282Y homozygous | 1 in 200 | High | Variable — ~50% of males, ~20% of females develop clinical disease |
| C282Y / H63D compound heterozygous | 1 in 50 | Low–moderate | Usually mild; significant overload uncommon unless cofactors present (alcohol, metabolic syndrome) |
| H63D homozygous | 1 in 50 | Very low | Minimal clinical significance in isolation |
| C282Y heterozygous | 1 in 8 | Negligible | Carrier state; no clinical iron overload expected |
Non-HFE Haemochromatosis
Rare non-HFE forms present with more aggressive iron loading, typically in younger patients:
- Juvenile haemochromatosis (Type 2): Mutations in HJV (hemojuvelin, Type 2A) or HAMP (hepcidin, Type 2B). Presents in the 2nd–3rd decade with severe cardiomyopathy, hypogonadism, and hepatic iron overload. Ferritin often >10,000 µg/L.
- TFR2-related haemochromatosis (Type 3): Mutations in transferrin receptor 2. Phenotype similar to HFE-HH but can present earlier. Autosomal recessive.
- Ferroportin disease (Type 4): Mutations in SLC40A1 (ferroportin). Two subtypes: loss-of-function (autosomal dominant, iron loading in macrophages, raised ferritin with lower transferrin saturation) and gain-of-function (resembles classic HH). Phlebotomy may be poorly tolerated due to anaemia in the loss-of-function variant.
Diagnostic Workup
The diagnostic approach follows a stepwise algorithm:
Investigations Summary
Treatment
Therapeutic Phlebotomy
Therapeutic venesection remains the cornerstone and most effective treatment for hereditary haemochromatosis. The goal is to deplete excess iron stores and then maintain ferritin at a low-normal level lifelong.
Iron Chelation Therapy
Chelation is reserved for patients in whom therapeutic phlebotomy is contraindicated or poorly tolerated:
- Severe anaemia (Hb <100 g/L and cannot tolerate venesection)
- Heart failure or significant cardiomyopathy
- Poor venous access
- Non-HFE haemochromatosis with rapid iron re-accumulation
Dietary Advice
While dietary modification alone cannot treat iron overload, the following advice is recommended to minimise additional iron absorption:
- Avoid iron supplements and multivitamins containing iron.
- Avoid vitamin C supplements — ascorbic acid enhances non-haem iron absorption by 2–3 fold. Dietary vitamin C from fruit is acceptable in normal quantities.
- Limit red meat — reduce intake but complete avoidance is unnecessary.
- Limit alcohol — alcohol increases iron absorption and accelerates liver injury. Particularly avoid alcohol with meals.
- Avoid raw or undercooked shellfish — especially oysters. Vibrio vulnificus is an iron-dependent bacterium; HH patients are at markedly increased risk of fatal sepsis with Vibrio infection.
- Tea and coffee consumed with meals may reduce non-haem iron absorption.
First-Degree Relative Screening
Once an index case of HFE-related haemochromatosis is confirmed, cascade screening of all first-degree relatives (parents, siblings, children) is mandatory:
- HFE genotyping to identify homozygous or compound heterozygous relatives.
- Iron studies (ferritin, TSAT) at time of genetic testing.
- Genetic counselling should be offered before testing, particularly for minors.
- Children of a confirmed C282Y homozygote are obligate heterozygotes; full genotyping of the other parent is required to determine risk of homozygosity in offspring.
- Pre-symptomatic detection and early phlebotomy prevents all major complications.
Complications
Iron deposition in parenchymal organs leads to progressive tissue damage. The risk of complications correlates with the degree and duration of iron overload. Early phlebotomy before organ damage occurs can prevent all of the following complications.
Cirrhosis
Hepatic iron deposition causes progressive fibrosis and ultimately cirrhosis, which is the most serious hepatic complication. Once cirrhosis is established, it is irreversible even with adequate iron depletion. Risk factors for cirrhosis include:
- Ferritin persistently >1000 µg/L
- Concurrent alcohol excess (synergistic hepatotoxicity)
- Hepatitis B or C co-infection
- Duration of iron overload (late diagnosis)
Hepatocellular Carcinoma (HCC)
HCC risk is increased in HH patients with established cirrhosis. Importantly, HCC may also develop in non-cirrhotic livers with very high iron stores, though this is less common. Surveillance recommendations:
- 6-monthly surveillance with serum alpha-fetoprotein (AFP) and hepatic ultrasound for all patients with established cirrhosis.
- Consider HCC surveillance in non-cirrhotic patients with persistently elevated ferritin >1000 µg/L, particularly if other risk factors present (co-infection, alcohol excess).
- Surveillance should continue lifelong even after adequate iron depletion if cirrhosis has been established.
Diabetes Mellitus
Iron deposition in the pancreatic islet cells causes beta-cell damage and insulin deficiency. The term "bronze diabetes" reflects the combination of iron-related skin pigmentation and secondary diabetes. Management includes:
- Standard diabetes management per Australian guidelines (ADIPS/RACGP).
- Glycaemic control may improve with adequate iron depletion if detected early.
- If cirrhosis coexists, consider hepatogenic diabetes and adjust HbA1c interpretation accordingly.
Cardiomyopathy and Arrhythmia
Myocardial iron deposition is a feared complication, particularly in juvenile haemochromatosis and severe HFE-HH:
- Restrictive cardiomyopathy progressing to dilated cardiomyopathy and heart failure.
- Cardiac arrhythmias: Atrial fibrillation, ventricular ectopy, and potentially fatal ventricular tachycardia.
- Cardiac T2* MRI is the gold standard for myocardial iron quantification.
- Aggressive iron chelation is essential when cardiac iron overload is demonstrated. Combination therapy (deferoxamine + deferiprone) may be required.
- Cardiac involvement may be reversible with intensive chelation if detected before irreversible fibrosis.
Hypogonadism
Iron deposition in the anterior pituitary (particularly gonadotrophs) causes secondary hypogonadotropic hypogonadism:
- Males: Erectile dysfunction, decreased libido, testicular atrophy, loss of secondary sexual characteristics. Testosterone replacement may be required.
- Females: Amenorrhoea, infertility, premature menopause. Oestrogen/progesterone replacement if symptomatic.
- Other pituitary axes may be affected (growth hormone deficiency, hypothyroidism).
- Ferritin >2000 µg/L significantly increases hypogonadism risk.
Arthropathy
Iron-related arthropathy is a distinctive and common complication affecting up to 50% of patients with HH:
- Classic pattern: Involvement of the 2nd and 3rd metacarpophalangeal joints (MCPJs) — a clinically distinctive feature that should prompt consideration of HH.
- Radiographic finding: "Hooked" or "beak-like" osteophytes at the MCPJs, particularly the index and middle finger MCPJs.
- Chondrocalcinosis (pseudogout) is common — iron promotes calcium pyrophosphate crystal deposition.
- Other affected joints: wrists, knees, hips, and first metatarsophalangeal joints.
- Arthropathy is often not reversible with phlebotomy once established. Early detection and treatment is critical.
- Management: NSAIDs, intra-articular corticosteroids, physiotherapy. Phlebotomy does not improve established joint damage.
Skin Hyperpigmentation
Bronze-grey skin discolouration results from increased melanin and iron deposition in the skin:
- Most noticeable on the face, neck, extensor surfaces of forearms, lower legs, and genitalia.
- May improve with iron depletion, though permanent pigmentation can occur.
- Distinctive when combined with the other classic features (diabetes, cirrhosis, cardiomyopathy).
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(1):328-343.
- 2. European Association for the Study of the Liver (EASL). EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3-22.
- 3. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388(10045):706-716.
- 4. Olynyk JK, Cullen DJ, Aquilia S, Rossi E, Summerville L, Powell LW. A population-based study of the clinical expression of the hemochromatosis gene. N Engl J Med. 1999;341(10):718-724.
- 5. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358(3):221-230.
- 6. Royal Australian College of General Practitioners (RACGP). Red Book: Guidelines for preventive activities in general practice. 9th ed. East Melbourne: RACGP; 2016.
- 7. Australian Institute of Health and Welfare (AIHW). Iron deficiency and iron overload. Cat. no. PHE 246. Canberra: AIHW; 2020.
- 8. Brancaleone V, Wijmenga C, Adrian TE, et al. Genetic hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2020;159(2):393-408.
- 9. Sandhu K, Flintoff K, Chatfield MD, et al. Phenotypic analysis of hemochromatosis subtypes reveals that C282Y homozygotes and H63D compound heterozygotes have distinct clinical profiles. Blood Adv. 2020;4(15):3512-3521.
- 10. National Health and Medical Research Council (NHMRC). NHMRC statement on genetic testing and information for the diagnosis and management of haemochromatosis. Canberra: NHMRC; 2007.
- 11. St Pierre TG, Clark PR, Chua-anusorn W, et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood. 2005;105(2):855-861.
- 12. Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352(17):1769-1778.
- 13. Gurrin LC, Bertalli NA, Dalton GW, et al. HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity. Hepatology. 2009;50(1):94-101.
- 14. Australian Red Cross Lifeblood. Hereditary haemochromatosis — blood donation. Sydney: Lifeblood; 2023. Available from: www.lifeblood.com.au.