📋 Key Information Summary
- Pulmonary hypertension (PH) is defined by a mean pulmonary arterial pressure (mPAP) ≥20 mmHg at rest measured by right heart catheterisation (RHC) — echocardiography alone is insufficient for diagnosis.
- The WHO classification divides PH into five groups: Group 1 — Pulmonary arterial hypertension (PAH); Group 2 — Left heart disease; Group 3 — Lung disease/hypoxia; Group 4 — Chronic thromboembolic PH (CTEPH); Group 5 — Unclear/multifactorial mechanisms.
- Group 1 PAH has the most complex pharmacotherapy pathway and requires specialist management at a PH centre; all other groups focus primarily on treating the underlying cause.
- Vasoreactivity testing with inhaled nitric oxide (iNO) or IV adenosine is mandatory in Group 1 PAH — only vasoreactive responders should receive calcium channel blocker (CCB) therapy (long-term high-dose).
- Risk stratification using ESC/ERS guidelines determines treatment strategy: low risk → monotherapy or combination; intermediate risk → initial combination; high risk → initial triple combination or parenteral prostacyclin.
- Key PH biomarkers include BNP/NT-proBNP, troponin, uric acid, and cardiac MRI parameters; 6-minute walk test (6MWT) and WHO functional class (I–IV) are essential clinical measures.
- CTEPH is the only potentially curable form of PH — all patients with CTEPH should be assessed for pulmonary endarterectomy (PEA) at an experienced centre; balloon pulmonary angioplasty (BPA) is for inoperable disease.
- Riociguat (Adempas®) is PBS-listed for inoperable/recurrent CTEPH; Bosentan (Tracleer®), sildenafil (Revatio®), ambrisentan (Volibris®), and treprostinil are PBS-listed for PAH under Authority Required arrangements.
- Pregnancy in PAH carries maternal mortality of 30–50% — pregnancy is strongly contraindicated; ERA-class drugs are teratogenic and require reliable contraception.
- Aboriginal and Torres Strait Islander Australians have higher rates of rheumatic heart disease–related PH and later presentations; culturally safe screening and access to specialist services require specific strategies.
- Referral to a specialist PH centre (e.g., Royal Melbourne, Royal Prince Alfred, St Vincent's Sydney, Prince Charles Brisbane) is recommended for all confirmed PAH and CTEPH cases.
Introduction & Australian Epidemiology
Pulmonary hypertension (PH) encompasses a heterogeneous group of conditions characterised by elevated pulmonary arterial pressure leading to right ventricular failure and death if untreated. The haemodynamic definition requires a mean pulmonary arterial pressure (mPAP) ≥20 mmHg at rest measured by right heart catheterisation (RHC), updated from the prior 25 mmHg threshold at the 6th World Symposium on Pulmonary Hypertension (2018).
In Australia, pulmonary arterial hypertension (PAH, WHO Group 1) has an estimated prevalence of approximately 15–50 per million population. Idiopathic PAH (IPAH) remains the most common subtype, with female predominance (2–4:1 F:M ratio). Associated forms include connective tissue disease–associated PAH (particularly systemic sclerosis), congenital heart disease–related PAH, and portopulmonary hypertension.
The Australian and New Zealand Society of Cardiac and Thoracic Surgeons (ANZSCTS) and the Thoracic Society of Australia and New Zealand (TSANZ) recommend referral to designated PH centres for all suspected PAH and CTEPH cases. Major PH centres in Australia include the Royal Melbourne Hospital PH Service, Royal Prince Alfred Hospital (Sydney), St Vincent's Hospital (Sydney), The Prince Charles Hospital (Brisbane), and Royal Adelaide Hospital.
The most common form of PH overall is Group 2 (left heart disease–related PH), which complicates up to 60–70% of patients with heart failure with preserved ejection fraction (HFpEF) and a significant proportion of those with heart failure with reduced ejection fraction (HFrEF). Group 3 (lung disease–related PH) is increasingly recognised in COPD and interstitial lung disease (ILD), particularly idiopathic pulmonary fibrosis (IPF).
CTEPH (Group 4) occurs in approximately 2–4% of patients following acute pulmonary embolism and is potentially curable with pulmonary endarterectomy. Balloon pulmonary angioplasty (BPA) has emerged as an option for inoperable CTEPH, now performed at several Australian centres.
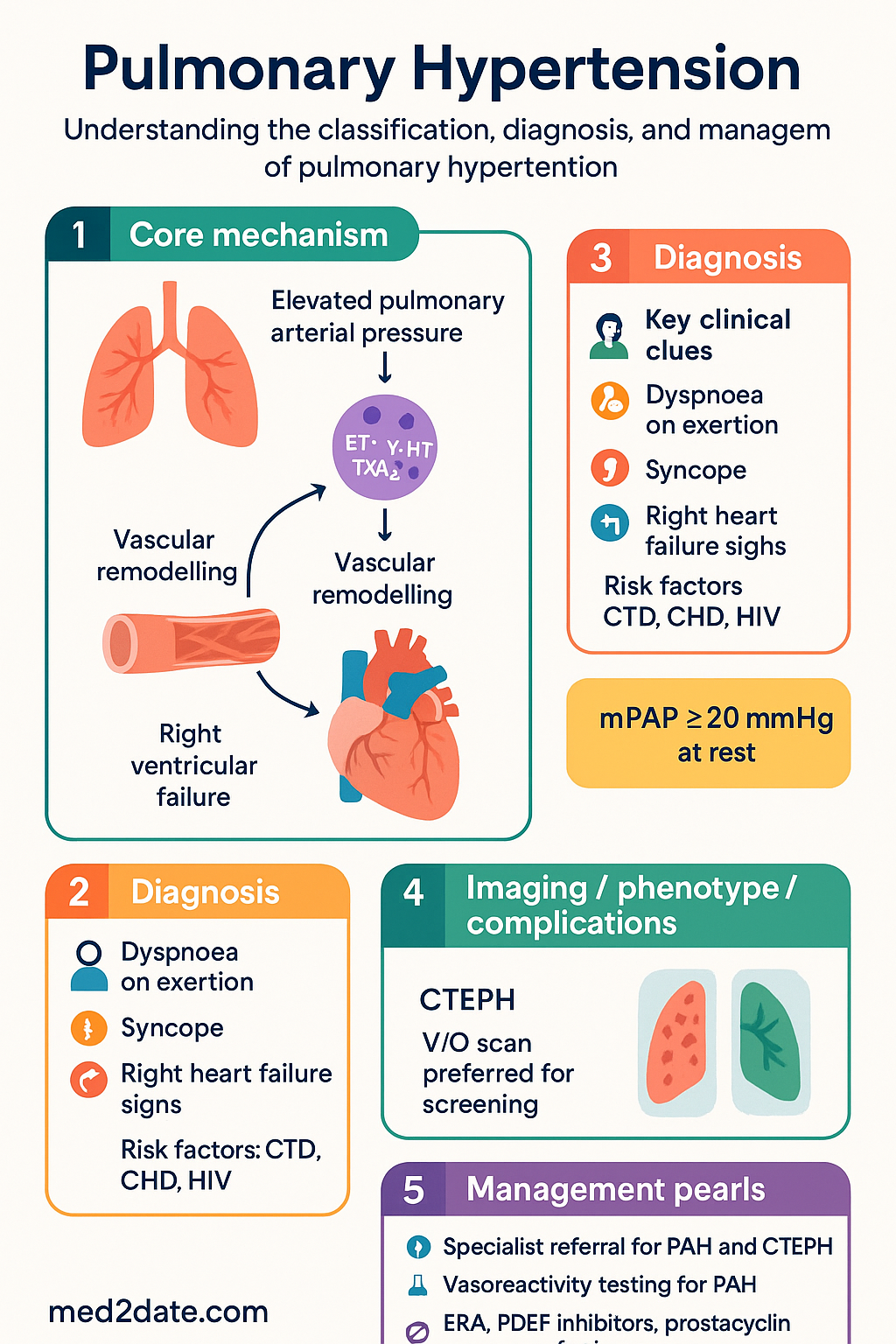
Classification & Diagnosis
WHO Haemodynamic Classification
The 6th World Symposium on Pulmonary Hypertension (Nice 2018) refined the haemodynamic definitions. PH is classified based on RHC findings into:
| Haemodynamic Definition | Mean PAP | PCWP | PVR |
|---|---|---|---|
| Pre-capillary PH (Groups 1, 3, 4, 5) | ≥20 mmHg | ≤15 mmHg | >3 WU |
| Post-capillary PH (Group 2) | ≥20 mmHg | >15 mmHg | ≤3 WU |
| Combined pre- and post-capillary PH | ≥20 mmHg | >15 mmHg | >3 WU |
PAP = pulmonary arterial pressure; PCWP = pulmonary capillary wedge pressure; PVR = pulmonary vascular resistance; WU = Wood units
WHO Clinical Classification (Groups 1–5)
The clinical classification system groups PH by shared pathophysiology, histopathology, and management approach:
| Group | Category | Examples |
|---|---|---|
| Group 1 | Pulmonary arterial hypertension (PAH) | Idiopathic (IPAH), heritable (BMPR2), drug/toxin-induced (anorexigens, methamphetamine), associated with CTD, HIV, portal hypertension, CHD, schistosomiasis |
| Group 1′ | Pulmonary veno-occlusive disease (PVOD) / Pulmonary capillary haemangiomatosis (PCH) | Rare; often heritable (EIF2AK4 mutations); poorly responsive to PAH-specific therapy |
| Group 1″ | Persistent PH of the newborn | Failure of postnatal pulmonary vascular remodelling |
| Group 2 | PH due to left heart disease | HFrEF, HFpEF, valvular disease (mitral/aortic), congenital/acquired left heart inflow/outflow tract obstruction |
| Group 3 | PH due to lung disease and/or hypoxia | COPD, ILD/IPF, combined pulmonary fibrosis and emphysema (CPFE), OHS, OSA, developmental lung disorders |
| Group 4 | PH due to pulmonary artery obstructions | Chronic thromboembolic pulmonary hypertension (CTEPH), other pulmonary artery obstructions (angiosarcoma, arteritis) |
| Group 5 | PH with unclear and/or multifactorial mechanisms | Haematological (chronic haemolytic anaemia, myeloproliferative), systemic (sarcoidosis, LCH, NF1), metabolic (Gaucher, glycogen storage), chronic renal failure on dialysis |
Diagnostic Algorithm
The diagnostic workup follows a stepwise approach beginning with clinical suspicion based on symptoms (progressive dyspnoea, exertional syncope, chest pain, fatigue) and risk factors:
Echocardiographic Screening — Probability of PH
The ESC/ERS echocardiographic probability assessment integrates multiple parameters:
| Probability | Peak TR Velocity (m/s) | Additional Echo Criteria |
|---|---|---|
| Low | ≤2.8 m/s or not measurable | No other PH signs |
| Intermediate | 2.9–3.4 m/s | Presence of ≥1 additional PH sign (RV dilation, RA enlargement, septal flattening, RVOT acceleration time <105 ms, elevated IVC) |
| High | >3.4 m/s | Regardless of additional signs |
Right Heart Catheterisation
RHC is mandatory before initiating PAH-specific therapy and provides critical haemodynamic data:
- Measured parameters: mPAP, systolic/diastolic PAP, PCWP (or left ventricular end-diastolic pressure), cardiac output (thermodilution or Fick), right atrial pressure (RAP), mixed venous oxygen saturation (SvO₂)
- Calculated parameters: PVR = (mPAP − PCWP) / CO; cardiac index (CI = CO/BSA); transpulmonary gradient (TPG = mPAP − PCWP); diastolic pressure gradient (DPG = dPAP − PCWP)
- Complications: Serious adverse events occur in <1% of procedures at experienced centres; include cardiac arrhythmia, vascular injury, pneumothorax, pulmonary artery rupture (rare)
- Frequency of follow-up RHC: Typically 3–6 months after initiating therapy, then annually or as clinically indicated to guide treatment decisions
Group 1 PAH — Management
General Measures & Supportive Therapy
- Supervised exercise/rehabilitation: Improves exercise capacity, functional class, and quality of life; should be offered at PH centres under specialist supervision
- Diuretics: Loop diuretics (furosemide, bumetanide) for fluid overload and right heart failure; spironolactone commonly added
- Anticoagulation: Controversial in IPAH; consider in IPAH and anorexigen-associated PAH (warfarin, target INR 1.5–2.5); NOT recommended in CTD-PAH or CHD-PAH
- Long-term oxygen therapy: If hypoxaemic (PaO₂ <60 mmHg at rest or on exertion)
- Pregnancy avoidance: ERA-class drugs are teratogenic; reliable contraception mandatory; pregnancy carries 30–50% maternal mortality in PAH
Vasoreactivity Testing & Calcium Channel Blockers
Vasoreactivity testing is performed during RHC in all suspected Group 1 PAH patients. A positive response is defined as a reduction in mPAP ≥10 mmHg to a value ≤40 mmHg with maintained or increased cardiac output.
PAH-Specific Pharmacotherapy Pathways
PAH-specific therapy targets three pathogenic pathways: the endothelin pathway (ERAs), the nitric oxide–cGMP pathway (PDE5 inhibitors, sGC stimulators), and the prostacyclin pathway (prostacyclin analogues, IP receptor agonists). Current ESC/ERS guidelines recommend risk-stratified initial combination therapy.
Endothelin Receptor Antagonists (ERAs)
PDE5 Inhibitors / sGC Stimulators (Nitric Oxide Pathway)
Prostacyclin Pathway Agents
Combination Therapy Strategy (ESC/ERS 2022)
Risk Stratification
Risk stratification is central to PAH management and determines treatment strategy, escalation timing, and transplant referral. The ESC/ERS guidelines employ a multi-parameter approach assessing 1-year mortality risk as low (<5%), intermediate (5–10%), or high (>10%).
ESC/ERS Risk Assessment Table
| Parameter | Low Risk (<5%) | Intermediate (5–10%) | High Risk (>10%) |
|---|---|---|---|
| WHO Functional Class | I, II | III | IV |
| 6MWT (metres) | >440 m | 165–440 m | <165 m |
| Cardiopulmonary exercise testing (peak VO₂) | >15 mL/min/kg | 11–15 mL/min/kg | <11 mL/min/kg |
| BNP / NT-proBNP | BNP <50; NT-proBNP <300 ng/L | BNP 50–300; NT-proBNP 300–1400 ng/L | BNP >300; NT-proBNP >1400 ng/L |
| Echocardiographic signs | No RA dilation, no pericardial effusion | RA moderate dilation, minimal effusion | Significant RA dilation, moderate–large pericardial effusion |
| Right atrial pressure (RHC) | <8 mmHg | 8–14 mmHg | >14 mmHg |
| Cardiac index (RHC) | ≥2.5 L/min/m² | 2.0–2.4 L/min/m² | <2.0 L/min/m² |
| SvO₂ (RHC) | >65% | 60–65% | <60% |
REVEAL Risk Score
The REVEAL (Registry to Evaluate Early and Long-term PAH Disease Management) score is a validated US-derived composite scoring system incorporating 12–13 variables to estimate 1-year survival. It has been incorporated into the 2022 ESC/ERS guidelines as a complementary tool:
| REVEAL Variable | Points |
|---|---|
| Male >60 years | +2 |
| PAH aetiology — scleroderma, portal, congenital (associated higher risk) | Varies (0–4) |
| Family history of PAH or BMPR2+ | +2 |
| NYHA/WHO FC IV | +3 |
| 6MWT <165 m | +2 |
| BNP >180 pg/mL or NT-proBNP >1500 pg/mL | +2 |
| Mean RAP >14 mmHg | +1 |
| PVR >32 WU | +1 |
| DLCO <40% predicted | +1 |
| Pericardial effusion on echo | +1 |
Low risk: REVEAL score ≤6 (≥95% 1-year survival); Intermediate: 7–8 (90–95%); High risk: ≥9 (<90% 1-year survival). The online calculator is available at revealcalc.hexacath.com.
Biomarkers in PH
| Biomarker | Significance | Available in Australia |
|---|---|---|
| BNP | RV wall stress; prognostic; serial monitoring guides treatment; most widely used PH biomarker | Yes — MBS item 66808; widely available |
| NT-proBNP | Similar prognostic utility to BNP; longer half-life; influenced by renal function | Yes — MBS item 66809 |
| High-sensitivity troponin | RV micro-injury; elevated in decompensated PAH; adverse prognostic marker | Yes — MBS item 66818 |
| Uric acid | Indirect marker of tissue hypoxia and impaired cardiac output; limited specificity | Yes — widely available |
| Cardiac MRI parameters | RVEF, RV volumes, late gadolinium enhancement (LGE); most accurate for RV function assessment; emerging prognostic value | Yes — MBS items for cardiac MRI; requires specialist referral |
6-Minute Walk Test (6MWT)
- Standardised submaximal exercise test — distance, SpO₂, heart rate, Borg dyspnoea score, and blood pressure recorded
- Normal distance: approximately 400–700 m (age and sex dependent); a distance <440 m correlates with intermediate risk; <165 m with high risk
- Serial 6MWT used to track treatment response — a decline of >30–50 m is clinically significant
- SpO₂ desaturation >10% or nadir <85% during 6MWT is an adverse prognostic sign
- Performed at baseline, 3–6 months after treatment initiation, then every 3–6 months in stable patients
Treatment Goals & Targets
Group 2–5 Management
Unlike Group 1 PAH, the primary therapeutic strategy for Groups 2–5 PH is identification and treatment of the underlying disease. PAH-specific therapies (ERAs, PDE5 inhibitors, prostacyclins) are generally NOT indicated in Group 2–3 PH and may cause harm. Exceptions exist in advanced cases referred to PH centres.
Group 2 — PH Due to Left Heart Disease
Group 2 is the most common cause of PH globally. The haemodynamic hallmark is post-capillary PH (PCWP >15 mmHg), which may be isolated (IpcPH) or combined with a pre-capillary component (CpcPH, PVR >3 WU). CpcPH carries worse prognosis.
| Underlying Condition | Management Strategy |
|---|---|
| HFrEF | GDMT for heart failure (ACEi/ARB/ARNI, beta-blocker, MRA, SGLT2i); device therapy (ICD/CRT); LVAD/transplant if advanced; treat iron deficiency (IV ferric carboxymaltose) |
| HFpEF | SGLT2i (dapagliflozin, empagliflozin — PBS-listed); diuretics for congestion; treat AF, hypertension, obesity, OSA; spironolactone; exercise training |
| Valvular disease | Timely surgical or percutaneous intervention (mitral clip, TAVI); management of mitral regurgitation, aortic stenosis; prosthetic valve assessment |
| Constrictive pericarditis | Pericardiectomy; echocardiography and CT/MRI assessment; specialist referral |
| Left-to-right shunts (ASD, VSD, PDA) | Defect closure if Qp:Qs >1.5 and PVR <5 WU; Eisenmenger syndrome — PAH-specific therapy may be considered at specialist PH/CHD centre |
Group 3 — PH Due to Lung Disease and/or Hypoxia
PH complicating lung disease is classified as mild (mPAP 21–24 mmHg), moderate (25–34 mmHg), or severe (≥35 mmHg). Severe PH in Group 3 is termed "severe PH-ILD" or "severe PH-COPD" and may be considered for referral to a PH centre.
| Underlying Condition | Management Strategy |
|---|---|
| COPD | GOLD guideline–directed therapy; LTOT if PaO₂ ≤55 mmHg; optimise inhaler therapy; pulmonary rehabilitation; smoking cessation (varenicline, NRT — PBS-listed) |
| ILD / IPF | Antifibrotic therapy (nintedanib, pirfenidone); LTOT; consider transplant assessment; PH in IPF — poor prognosis; inhaled treprostinil studied in PH-ILD (INCREASE trial) |
| CPFE | Combined emphysema + fibrosis; disproportionate PH; LTOT; transplant referral; high risk of lung cancer |
| Obesity hypoventilation syndrome (OHS) | NIV/CPAP; weight management (GLP-1 receptor agonists if eligible); treat OSA |
| OSA | CPAP — PH typically mild and improves with CPAP compliance; reassess PH 3–6 months after CPAP initiation |
Group 5 — Unclear / Multifactorial Mechanisms
Group 5 PH encompasses conditions with incompletely understood pathophysiology or multifactorial mechanisms. Management focuses on treating the underlying condition:
| Subgroup | Management |
|---|---|
| Haematological (chronic haemolytic anaemia, thalassaemia, SCD, myeloproliferative) | Optimise disease-specific therapy; exchange transfusion in SCD; hydroxyurea; specialist haematology input; PH-specific therapy may be considered in refractory cases |
| Systemic disorders (sarcoidosis, LCH, NF1, vasculitis) | Immunosuppressive therapy for active sarcoidosis; PH-specific therapy in refractory cases at PH centre; corticosteroids, methotrexate, azathioprine |
| Metabolic (Gaucher, glycogen storage diseases, thyroid disease) | Enzyme replacement for Gaucher; treat thyroid dysfunction; manage glycogen storage disease with specialist metabolic team |
| Chronic kidney disease / dialysis | Optimise dialysis; manage fluid overload; treat AV fistula–related high-output state; renal transplant if eligible |
CTEPH Management (Group 4)
Chronic thromboembolic pulmonary hypertension (CTEPH) is characterised by organised thrombus in the pulmonary arteries leading to increased PVR and PH. It is the only potentially curable form of PH. CTEPH develops in approximately 2–4% of patients within 2 years following acute pulmonary embolism, though up to 40% of CTEPH patients have no documented prior PE.
CTEPH Diagnostic Criteria
- mPAP ≥25 mmHg at rest by RHC (note: this higher threshold than general PH is maintained for CTEPH to require haemodynamic significance)
- PCWP ≤15 mmHg (pre-capillary pattern)
- Evidence of chronic thromboembolic disease on imaging (CTPA, pulmonary angiography, or MRA) — ring-like stenosis, webs, bands, complete vascular occlusions
- Adequate anticoagulation for ≥3 months
Pulmonary Endarterectomy (PEA)
PEA is the treatment of choice for operable CTEPH and offers potential cure by restoring pulmonary vascular patency. Operability assessment requires multidisciplinary team discussion (MDT) involving thoracic/cardiac surgeons, PH physicians, and radiologists.
Balloon Pulmonary Angioplasty (BPA)
BPA is an interventional radiology procedure for patients with inoperable CTEPH or residual/recurrent PH after PEA. It involves staged dilation of segmental and subsegmental pulmonary arteries using balloon catheters under fluoroscopic guidance.
- Indication: Inoperable CTEPH (distal disease, significant comorbidities, patient refusal of surgery, or residual PH post-PEA)
- Procedure: Performed in 4–8 sessions at 4–6-week intervals; each session targets 1–3 segments
- Outcomes: Improved mPAP, PVR, 6MWT, and WHO FC; 5-year survival >90% in experienced centres; CTEPH centres in Australia performing BPA include Royal Prince Alfred and The Prince Charles Hospital
- Complications: Reperfusion pulmonary oedema (10–15%), pulmonary artery injury/perforation (1–2%), haemoptysis
Medical Therapy for Inoperable CTEPH
For patients with inoperable CTEPH or persistent/recurrent PH after PEA/BPA, targeted medical therapy is indicated:
Additional agents that may be used off-label or in combination at specialist PH centres include bosentan (BENEFIT trial showed some benefit in CTEPH), macitentan, sildenafil, and treprostinil. All require specialist oversight.
Lifelong Anticoagulation
All CTEPH patients require lifelong anticoagulation to prevent recurrent thromboembolism:
- Warfarin (target INR 2.0–3.0) — traditionally preferred; required for patients on riociguat due to limited DOAC data in CTEPH
- DOACs: Edoxaban, rivaroxaban used increasingly; limited CTEPH-specific data but extrapolated from PE treatment; discuss with PH specialist
- Inferior vena cava (IVC) filter: Consider in patients with recurrent VTE despite anticoagulation or absolute contraindication to anticoagulation
Special Populations
Pregnancy
Paediatrics
Elderly
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal and Torres Strait Islander Health Considerations
Aboriginal and Torres Strait Islander Australians experience a disproportionate burden of conditions leading to pulmonary hypertension, yet face significant barriers to timely diagnosis, specialist access, and treatment. Rheumatic heart disease (RHD), a preventable condition virtually eliminated in non-Indigenous Australia, remains a leading cause of Group 2 PH in remote Aboriginal and Torres Strait Islander communities, particularly in the Northern Territory, Far North Queensland, and Western Australia.
Key Epidemiological Disparities
- Rheumatic heart disease (RHD): Aboriginal and Torres Strait Islander Australians have among the highest RHD rates globally (incidence up to 150/100,000 in NT children); RHD leads to significant valvular disease and subsequent Group 2 PH
- Chronic lung disease: Higher rates of COPD, bronchiectasis, and post-infectious lung disease contributing to Group 3 PH; smoking prevalence remains high (approximately 40% in Aboriginal and Torres Strait Islander adults vs 11% non-Indigenous)
- Later presentation: Aboriginal and Torres Strait Islander patients with PH present at more advanced stages due to healthcare access barriers and delayed referral pathways
- Congenital heart disease: Higher prevalence of undiagnosed congenital heart disease contributing to PAH in remote communities
📚 References
- 1. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Heart J. 2016;37(1):67–119.
- 2. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618–3731.
- 3. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913.
- 4. Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–172.
- 5. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–2533.
- 6. Ghofrani HA, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–340.
- 7. Galiè N, Barberà JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834–844.
- 8. Jenkins D, Madani M, Fadel E, et al. Pulmonary endarterectomy in the management of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2017;26(143):160111.
- 9. Ogawa A, Satoh T, Fukuda T, et al. Balloon pulmonary angioplasty for chronic thromboembolic pulmonary hypertension: results of a multicenter registry. Circ Cardiovasc Qual Outcomes. 2017;10(11):e004029.
- 10. Australian Institute of Health and Welfare (AIHW). Rheumatic heart disease and acute rheumatic fever in Australia: 2017–2022. Cat. No. CVD 92. Canberra: AIHW; 2023.
- 11. RHDAustralia (ARF/RHD writing group). Australian guideline for prevention, diagnosis and management of acute rheumatic fever and rheumatic heart disease. 3rd ed. Darwin: Menzies School of Health Research; 2024.
- 12. Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894–2903.
- 13. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. J Am Coll Cardiol. 2009;53(17):1573–1619.
- 14. National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand. Guidelines for the prevention, detection and management of chronic heart failure in Australia. Updated 2024.
- 15. Thenappan T, Ormiston ML, Ryan JJ, et al. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492.