📋 Key Information Summary
- Interstitial lung diseases (ILDs) encompass over 200 distinct entities; a systematic approach to classification using HRCT patterns, multidisciplinary discussion (MDD), and selective lung biopsy is essential for accurate diagnosis.
- Idiopathic pulmonary fibrosis (IPF) is the most common and lethal idiopathic ILD; the hallmark is a usual interstitial pneumonia (UIP) pattern on HRCT — honeycombing, traction bronchiectasis, and subpleural basal predominance with no inconsistent features.
- Antifibrotic therapy with nintedanib (Ofev®) or pirfenidone (Esbriet®) slows FVC decline in IPF; both are PBS-listed (Authority Required) and should be initiated early by a respiratory physician experienced in ILD.
- Acute exacerbations of IPF carry in-hospital mortality of 50–80%; empirical high-dose corticosteroids and supportive care in ICU are the mainstay — lung transplant referral should be considered before functional decline.
- Hypersensitivity pneumonitis (HP) requires identification and removal of the causative antigen; chronic HP may mimic IPF and is distinguished by upper/mid-zone predominance, mosaic attenuation, and air trapping on expiratory HRCT.
- Sarcoidosis staging (0–IV) is based on chest radiography; Stage I (bilateral hilar lymphadenopathy alone) has >60% spontaneous resolution, whereas Stage IV (pulmonary fibrosis) requires long-term management.
- Cardiac sarcoidosis (2–5% of cases) can cause sudden death; cardiac MRI with late gadolinium enhancement and FDG-PET are key investigations; all patients with suspected sarcoidosis should have a 12-lead ECG and Holter monitor.
- Connective tissue disease-associated ILD (CTD-ILD) is most common in systemic sclerosis (SSc-ILD), rheumatoid arthritis (RA-ILD), and inflammatory myositis; mycophenolate mofetil and rituximab are increasingly used as first-line steroid-sparing agents.
- In SSc-ILD, nintedanib (SENSCIS trial) is PBS-listed for slowing FVC decline; mycophenolate is the conventional first-line immunosuppressant per Australian consensus.
- Aboriginal and Torres Strait Islander Australians have higher rates of chronic lung disease, delayed access to specialist ILD care, and reduced access to lung transplant evaluation — proactive outreach and culturally safe care are critical.
- All patients with progressive ILD should be discussed at a dedicated ILD multidisciplinary meeting (pulmonology, radiology, pathology, rheumatology) — biopsy decisions, treatment escalation, and transplant referral should be MDD-driven.
- Avoid empirical immunosuppression in suspected IPF — the PANTHER-IPF trial demonstrated harm from triple therapy (prednisolone + azathioprine + N-acetylcysteine); antifibrotic therapy is the evidence-based approach.
Introduction & Australian Epidemiology
Interstitial lung disease (ILD) is an umbrella term for a heterogeneous group of over 200 parenchymal lung disorders characterised by varying degrees of inflammation and fibrosis of the pulmonary interstitium. These conditions share common presenting features — progressive exertional dyspnoea, non-productive cough, and diffuse bilateral infiltrates on imaging — but differ vastly in aetiology, prognosis, and management.
In Australia, ILD prevalence is estimated at 60–80 per 100,000 population, with idiopathic pulmonary fibrosis (IPF) accounting for approximately 25–35% of all ILD diagnoses. IPF incidence rises sharply with age, with a median age at diagnosis of 65–70 years and a male-to-female ratio of approximately 1.5:1. Median survival from diagnosis remains 3–5 years — worse than many cancers.
The Australian IPF Registry (AIPFR), coordinated through the Lung Foundation Australia, has provided critical data on Australian disease patterns, comorbidities, and treatment outcomes. Key Australian data show that the median time from symptom onset to diagnosis is 12–18 months, highlighting the need for greater awareness among primary care clinicians.
Hypersensitivity pneumonitis is an important ILD subtype in Australia due to occupational and environmental exposures including bird keeping (budgerigar fancier's lung), farming, and mould exposure in tropical climates. Sarcoidosis shows lower prevalence in Australian Indigenous populations compared with African-American populations in the United States, but remains under-recognised in Aboriginal and Torres Strait Islander communities.
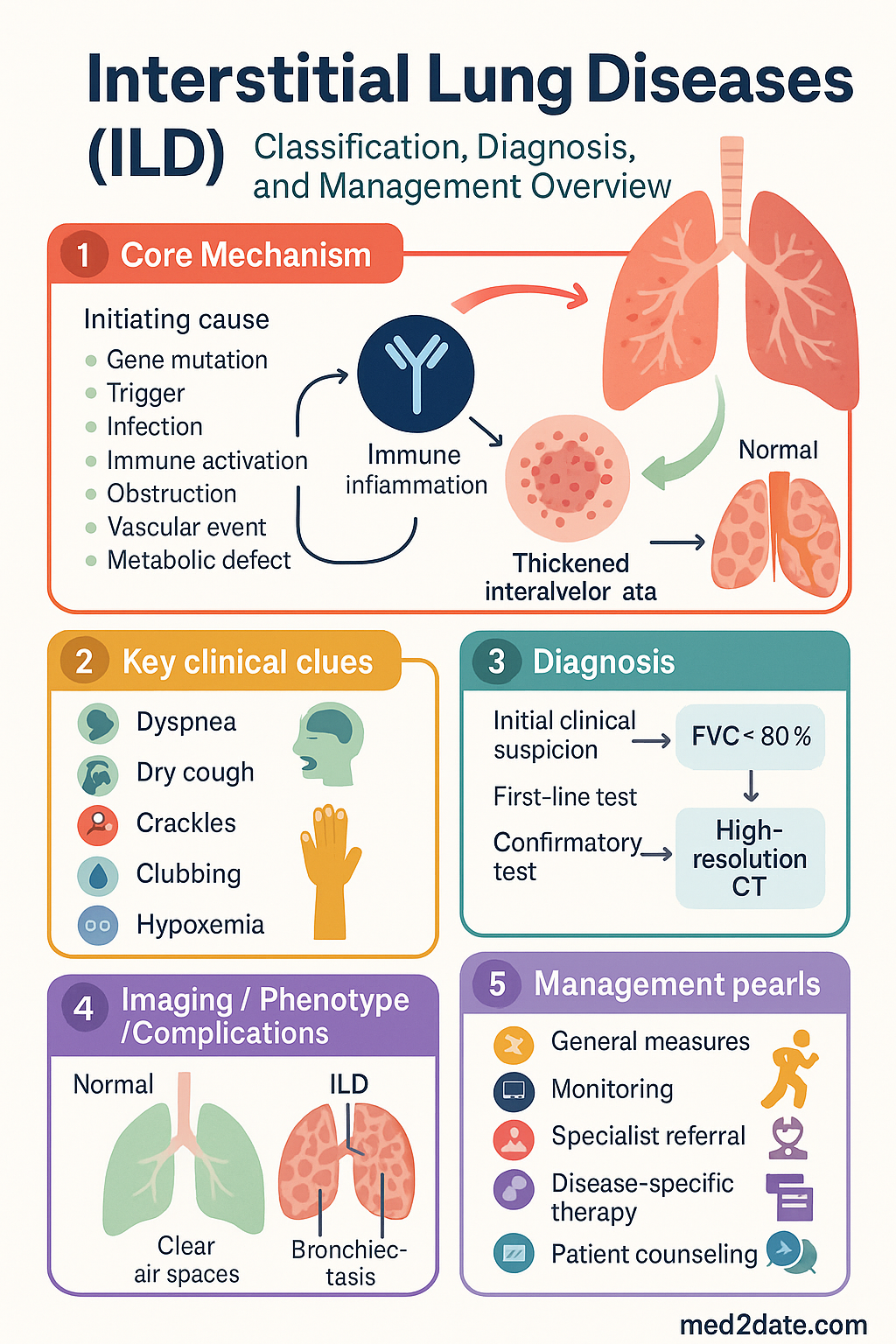
Classification & Diagnosis
Major ILD Subtypes
ILDs are broadly classified into those with known causes and idiopathic forms. The 2013 ATS/ERS classification, updated in 2018 and 2022, provides the framework used in Australian practice:
| Category | Examples | Key Features |
|---|---|---|
| Idiopathic interstitial pneumonias (IIPs) | IPF (UIP), NSIP, COP, AIP, DIP, RB-ILD, LIP | Diagnosis of exclusion; requires MDD |
| Connective tissue disease-associated | SSc-ILD, RA-ILD, myositis-ILD, SLE-ILD, Sjögren-ILD | Screen with autoimmune serology (ANA, RF, anti-CCP, myositis antibodies) |
| Hypersensitivity pneumonitis | Bird fancier's lung, farmer's lung, hot tub lung | Antigen identification critical; acute, subacute, chronic forms |
| Granulomatous | Sarcoidosis | Non-caseating granulomas; multi-organ involvement |
| Occupational / environmental | Asbestosis, silicosis, coal worker's pneumoconiosis | Exposure history essential; notifiable in some states |
| Drug-induced | Methotrexate, amiodarone, nitrofurantoin, checkpoint inhibitors | Temporal relationship to drug; may mimic any pattern |
| Smoking-related | RB-ILD, DIP, pulmonary Langerhans cell histiocytosis | Smoking cessation is first-line therapy |
HRCT Patterns and Diagnostic Significance
High-resolution computed tomography (HRCT) is the cornerstone of ILD diagnosis. A thin-section (1–1.5 mm) volumetric HRCT performed at full inspiration with expiratory images is the minimum standard in Australian centres. HRCT patterns are classified according to the 2018 ATS/ERS/JRS/ALAT guidelines:
| HRCT Pattern | Key Features | Primary Diagnosis |
|---|---|---|
| UIP (Usual Interstitial Pneumonia) | Subpleural, basal predominance; honeycombing ± traction bronchiectasis; minimal ground-glass; no inconsistent features | IPF (if no identifiable cause) |
| Probable UIP | Subpleural basal reticulation + traction bronchiectasis; no honeycombing; no inconsistent features | Likely IPF — MDD + consider biopsy |
| Indeterminate for UIP | Subpleural basal predominance but some atypical features | Surgical lung biopsy often required |
| NSIP pattern | Ground-glass opacity ± fine reticulation; subpleural sparing; basal predominance | CTD-ILD, idiopathic NSIP |
| OP (Organising Pneumonia) | Bilateral patchy consolidation, peribronchial; migratory; "reversed halo" sign | COP, secondary OP |
| HP pattern | Centrilobular nodules, mosaic attenuation, air trapping on expiration; upper/mid-zone predominance | Hypersensitivity pneumonitis |
Multidisciplinary Discussion (MDD)
Australian and international guidelines mandate that ILD diagnosis be made through multidisciplinary discussion involving a respiratory physician, thoracic radiologist, and lung pathologist, with rheumatology input when CTD-ILD is suspected. The MDD improves diagnostic accuracy from ~70% (individual clinician) to >90% and is recommended as best practice by the Thoracic Society of Australia and New Zealand (TSANZ).
Dedicated ILD MDD meetings are available at major Australian tertiary centres including Royal Melbourne Hospital, Alfred Health, Royal Brisbane and Women's Hospital, Royal Prince Alfred Hospital, and Fiona Stanley Hospital. Telemedicine MDD platforms are increasingly used to improve access for regional and remote Australian patients.
Lung Biopsy Indications
Idiopathic Pulmonary Fibrosis (IPF)
UIP Pattern on HRCT
The definitive HRCT diagnosis of IPF requires a UIP pattern (ATS/ERS 2018 update). The key features are:
- Typical UIP: Subpleural and basal predominant honeycombing with or without traction bronchiectasis, and minimal ground-glass opacity. No features inconsistent with UIP (upper/mid-zone or peribronchovascular predominance, extensive ground-glass, profuse micronodules, discrete cysts, mosaic attenuation with air trapping on three lobes, consolidation).
- Probable UIP: Subpleural basal reticular abnormality with traction bronchiectasis (or bronchiectasis) but no honeycombing and no inconsistent features. Lung biopsy may be needed for confirmation.
- Indeterminate: Some features of UIP but atypical findings present — surgical lung biopsy usually required.
- Alternative diagnosis: Features suggesting CTD-ILD, HP, drug effect, or another specific diagnosis.
Antifibrotic Therapy
Two antifibrotic agents are approved and PBS-listed in Australia for IPF. Neither reverses established fibrosis, but both significantly slow the rate of FVC decline — the primary endpoint in clinical trials.
Disease Progression and Monitoring
IPF follows a variable course. Disease progression is defined by:
- Forced vital capacity (FVC) decline ≥10% relative over 12 months — major prognostic indicator
- FVC decline 5–10% relative — marginal progression; warrants close monitoring
- DLCO decline ≥15% relative — associated with mortality increase
- Progression on HRCT — increasing extent of fibrosis, honeycombing
- Acute exacerbation — rapid worsening over <1 month with new bilateral ground-glass opacities; mortality 50–80%
Monitoring schedule: PFTs (spirometry + DLCO) every 3–6 months; 6-minute walk test (6MWT) every 6 months; HRCT annually or if clinical worsening; GAP index at diagnosis and annually.
| GAP Component | Points |
|---|---|
| Gender | Female = 0, Male = 1 |
| Age | <60 = 0, 60–65 = 1, >65 = 2 |
| Physiology (FVC % predicted) | >75% = 0, 50–75% = 1, <50% = 2 |
| Physiology (DLCO % predicted) | >55% = 0, 36–55% = 1, ≤35% = 2, cannot perform = 3 |
Interpretation: Stage I (0–3 points): median survival >5 years. Stage II (4–5 points): median survival 3–4 years. Stage III (6–8 points): median survival 1.5–2 years.
Lung Transplant Timing
IPF is the leading indication for lung transplantation in Australia. Referral should be made early — at the time of diagnosis or when any of the following are present:
- DLCO <40% predicted
- FVC decline ≥10% over 6 months
- Desaturation <88% on 6MWT
- Honeycombing on HRCT (UIP pattern)
- Hospitalisation for respiratory decline or acute exacerbation
- GAP score Stage II or III
Australian transplant centres: Alfred Health (Melbourne), St Vincent's Hospital (Sydney), Prince Charles Hospital (Brisbane), Royal Adelaide Hospital, Fiona Stanley Hospital (Perth). Median wait time in Australia is 6–18 months; living-donor lobar transplantation is occasionally performed. Contraindications include active malignancy, active infection, uncontrolled psychiatric illness, BMI >35, and non-adherence.
Hypersensitivity Pneumonitis
Antigen Identification
Hypersensitivity pneumonitis (HP) is an immunologically mediated inflammatory and/or fibrotic lung disease caused by inhalation of a causative antigen in a sensitised individual. In Australia, the most common exposures include:
| Exposure | Common Name | Antigen Source |
|---|---|---|
| Avian proteins | Budgerigar fancier's / pigeon fancier's lung | Bird droppings, feathers, serum proteins |
| Farming / agriculture | Farmer's lung | Thermoactinomyces, Aspergillus spp. in mouldy hay |
| Hot tub / spa | Hot tub lung | Mycobacterium avium complex in poorly maintained water |
| Mould exposure | Humidifier lung / tropical HP | Aspergillus, Cladosporium, Trichosporon |
| Chemical / industrial | Isocyanate HP | Paints, polyurethane foam — occupational (WorkSafe) |
A thorough occupational and environmental history is essential. Precipitating antibodies (IgG panels to avian antigens, moulds) support but do not confirm the diagnosis — positive serology is found in up to 50% of exposed but asymptomatic individuals. Bronchoalveolar lavage (BAL) showing lymphocytosis with a CD4:CD8 ratio <1 (or <2 with very high lymphocyte count) is characteristic but not pathognomonic.
Acute vs Chronic Hypersensitivity Pneumonitis
Steroid Therapy
Corticosteroids are the mainstay of treatment for subacute and chronic HP when antigen avoidance alone is insufficient:
Antigen Avoidance Strategies
Complete antigen avoidance is the most effective intervention and the only intervention that can halt disease progression. Strategies include:
- Remove birds from the home; bird cages, feathers, and droppings from living areas. Rehoming birds may be necessary.
- For occupational exposures: use P2/N95 respirators, improve ventilation, modify work practices, consider role change if severe disease.
- For mould-related HP: address water damage, improve ventilation, decontaminate HVAC systems; environmental assessment by occupational hygienist.
- Hot tub: drain, thoroughly clean, and ensure appropriate chlorination/bromination; consider discontinuation.
- Workers' compensation and WorkSafe claims should be initiated for occupationally acquired HP (varies by state/territory).
Sarcoidosis
Staging (Chest Radiograph — Scadding Stage)
Extrapulmonary Manifestations
Sarcoidosis is a systemic disease affecting virtually any organ. Australian data suggest extrapulmonary involvement in 30–50% of patients. Key manifestations include:
| Organ System | Manifestation | Frequency |
|---|---|---|
| Skin | Erythema nodosum, lupus pernio, maculopapular lesions | 20–35% |
| Eyes | Anterior uveitis (most common), posterior uveitis, retinal vasculitis | 10–25% |
| Cardiac | Conduction blocks, ventricular arrhythmias, cardiomyopathy, sudden death | 2–5% clinically; up to 25% subclinical at autopsy |
| Neurological | Cranial neuropathy (CN VII palsy), aseptic meningitis, peripheral neuropathy, pituitary involvement | 5–15% |
| Musculoskeletal | Acute arthritis (Löfgren syndrome), chronic bone cysts, myopathy | 10–15% |
| Renal | Hypercalcaemia/hypercalciuria (1α-hydroxylase), nephrocalcinosis | 5–10% |
| Liver | Granulomatous hepatitis, hepatomegaly | 10–20% |
Treatment Indications
Not all sarcoidosis requires treatment. Treatment is indicated when there is:
- Significant or progressive pulmonary involvement (declining FVC, worsening HRCT)
- Cardiac sarcoidosis (any evidence — treat regardless of symptoms)
- Neurosarcoidosis (any evidence — treat urgently)
- Severe ocular disease unresponsive to topical therapy
- Disfiguring skin disease (lupus pernio)
- Hypercalcaemia causing renal impairment
- Symptomatic systemic disease affecting quality of life
Immunosuppression
Cardiac Sarcoidosis
Neurosarcoidosis
Neurosarcoidosis requires urgent specialist management. Presentation includes cranial nerve palsies (especially facial nerve CN VII), aseptic meningitis, peripheral neuropathy, hypothalamic/pituitary dysfunction (diabetes insipidus), and spinal cord involvement. Diagnosis is supported by MRI brain with gadolinium (leptomeningeal enhancement), CSF analysis (elevated protein, lymphocytic pleocytosis, elevated ACE), and biopsy when accessible. Treatment: high-dose oral prednisolone (1 mg/kg/day) or IV methylprednisolone (1 g daily for 3–5 days), followed by steroid-sparing agents (methotrexate, mycophenolate, or infliximab for refractory cases).
Connective Tissue Disease-Associated ILD (CTD-ILD)
ILD is a significant cause of morbidity and mortality in connective tissue diseases (CTDs). Screening for ILD is recommended at CTD diagnosis and periodically thereafter, particularly in high-risk populations. The three most important CTD-ILD associations in Australian practice are rheumatoid arthritis, systemic sclerosis, and inflammatory myositis.
Rheumatoid Arthritis-Associated ILD (RA-ILD)
- Prevalence: 5–10% of RA patients (HRCT-detected subclinical ILD may be as high as 20–30%)
- Risk factors: male sex, smoking history, high-titre RF/anti-CCP, older age at RA diagnosis, usual interstitial pneumonia (UIP) pattern
- Most common pattern: UIP (60–70%), followed by NSIP
- RA-ILD with UIP pattern has a prognosis similar to IPF (median survival 3–5 years)
- Methotrexate pneumonitis is a diagnosis of exclusion — do not assume methotrexate is the cause without MDD review; true methotrexate ILD is rare (<1%)
- Screen with HRCT at RA diagnosis if risk factors present; PFTs (spirometry + DLCO) annually
Systemic Sclerosis-Associated ILD (SSc-ILD)
SSc-ILD is the leading cause of death in systemic sclerosis. ILD prevalence is 40–80% on HRCT (depending on disease subtype — diffuse cutaneous SSc has higher risk than limited cutaneous SSc). Key Australian management principles:
- Baseline HRCT and PFTs at SSc diagnosis — all patients, regardless of respiratory symptoms
- Indicators of progressive ILD: Extent of ILD on HRCT >20%, FVC <70% predicted, FVC decline ≥10% over 12 months, diffuse cutaneous subtype, anti-Scl-70 (anti-topoisomerase I) antibody positivity
- First-line immunosuppression: Mycophenolate mofetil 1000–1500 mg PO BD (SLS I trial, Australian consensus)
- Nintedanib is PBS-listed (Authority Required) for SSc-ILD based on the SENSCIS trial, which demonstrated a 44% reduction in the annual rate of FVC decline
- Combination therapy (mycophenolate + nintedanib) may be considered for progressive SSc-ILD per SENSCIS post-hoc and INBUILD subgroup analyses
Myositis-Associated ILD
Inflammatory myositis (dermatomyositis, polymyositis, antisynthetase syndrome, immune-mediated necrotising myopathy) is complicated by ILD in 20–40% of cases. Myositis-associated ILD can be severe and rapidly progressive, particularly in the antisynthetase syndrome (anti-Jo-1, anti-PL-7, anti-PL-12, anti-EJ, anti-OJ antibodies) and anti-MDA5 antibody-positive clinically amyopathic dermatomyositis (which carries a risk of rapidly progressive ILD with high mortality).
Treatment of myositis-ILD follows a step-up approach:
- First-line: High-dose corticosteroids (prednisolone 1 mg/kg/day, wean over 6–12 months) + steroid-sparing agent (mycophenolate mofetil or azathioprine)
- Second-line / steroid-sparing: Rituximab (MabThera®) 1000 mg IV × 2 doses (day 1 and 15); evidence from RIM and MyoNet registries, increasingly used as early steroid-sparing strategy
- Refractory disease: IV immunoglobulin (IVIg) 2 g/kg over 2–5 days monthly; cyclophosphamide; calcineurin inhibitors (tacrolimus, cyclosporine); JAK inhibitors (tofacitinib) — emerging evidence
- Nintedanib may be added for progressive fibrotic CTD-ILD (INBUILD trial — included CTD-ILD subgroup)
Immunosuppressive Strategies — Summary
| CTD-ILD Subtype | First-Line | Second-Line | Antifibrotic Role |
|---|---|---|---|
| SSc-ILD | Mycophenolate | Rituximab, cyclophosphamide, tocilizumab | Nintedanib (PBS-listed); combination with MMF |
| RA-ILD | Mycophenolate, rituximab (if UIP pattern) | Azathioprine, abatacept (less ILD risk than other biologics) | Nintedanib (INBUILD subgroup data); pirfenidone (INJOURNEY data) |
| Myositis-ILD | Corticosteroids + mycophenolate | Rituximab, IVIg, calcineurin inhibitors, cyclophosphamide | Consider for fibrotic phenotype |
| Sjögren-ILD | Corticosteroids + mycophenolate | Rituximab, azathioprine | Limited data; consider for progressive fibrotic disease |
Pathophysiology
The pathophysiology of ILD varies by subtype but centres on three core mechanisms:
- Inflammation-driven: Inflammatory cells (lymphocytes, macrophages, neutrophils) infiltrate the alveolar walls and interstitium, causing alveolitis. This is the predominant mechanism in HP, sarcoidosis, and CTD-ILD. Immunosuppression can halt or reverse this process.
- Fibrosis-driven: Abnormal wound healing leads to myofibroblast proliferation, extracellular matrix deposition, and progressive scarring. Alveolar epithelial injury with aberrant repair is the central paradigm in IPF. TGF-β, PDGF, and FGF pathways are key mediators targeted by antifibrotic agents.
- Granulomatous: Non-caseating granulomas composed of epithelioid histiocytes, giant cells, and CD4+ T lymphocytes. The hallmark of sarcoidosis. Driven by exaggerated Th1 immune response.
In IPF, the current paradigm favours repeated micro-injury to the alveolar epithelium (genetic predisposition + environmental triggers such as smoking, dust, gastro-oesophageal reflux) leading to aberrant fibroblast activation and progressive fibrosis without significant preceding inflammation. This explains why immunosuppression is ineffective and potentially harmful in IPF.
Investigations
Monitoring
Special Populations
Pregnancy
Paediatrics
Elderly
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal and Torres Strait Islander Health Considerations
Quick Reference — Key Drug Regimens
📚 References
- 1. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(4):e18–e47.
- 2. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68.
- 3. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082.
- 4. King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092.
- 5. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019;380(26):2518–2528.
- 6. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–1727.
- 7. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–2666.
- 8. Tzouvelekis A, Bonella F, Molina-Molina M, et al. Diagnosis and management of idiopathic pulmonary fibrosis: Updated ATS/ERS/JRS/ALAT guideline. Eur Respir J. 2023;62(2):2300234.
- 9. Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–1977.
- 10. Grutters JC, du Bois RM. Genetics of fibrosing lung diseases. Eur Respir J. 2005;25(5):915–927.
- 11. Jo HE, Glaspole I, Grainge C, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian IPF Registry. Eur Respir J. 2017;49(1):1601592.
- 12. Vasakova M, Morell F, Walsh S, et al. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. Am J Respir Crit Care Med. 2017;196(6):680–689.
- 13. Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America: Analysis Based on Health Care Use. Ann Am Thorac Soc. 2016;13(8):1244–1252.
- 14. Australian Institute of Health and Welfare. Aboriginal and Torres Strait Islander Health Performance Framework. AIHW; 2023.
- 15. Lung Foundation Australia. Patient and Carer Support — Pulmonary Fibrosis. Brisbane: Lung Foundation Australia; 2023. Available at: lungfoundation.com.au.
- 16. Assayag D, Vittinghoff E, Ryerson CJ, et al. The effect of gender on survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(4):440–446.
- 17. Ryerson CJ, Vittinghoff E, Ley B, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723–728.
- 18. Thoracic Society of Australia and New Zealand (TSANZ). Position Statement on Interstitial Lung Disease Multidisciplinary Meetings. Sydney: TSANZ; 2021.