📋 Key Information Summary
- Alpha-1 antitrypsin deficiency (AATD) is an autosomal codominant genetic disorder caused by mutations in the SERPINA1 gene, leading to reduced circulating alpha-1 antitrypsin (AAT) and increased risk of premature emphysema, particularly in the lower lobes.
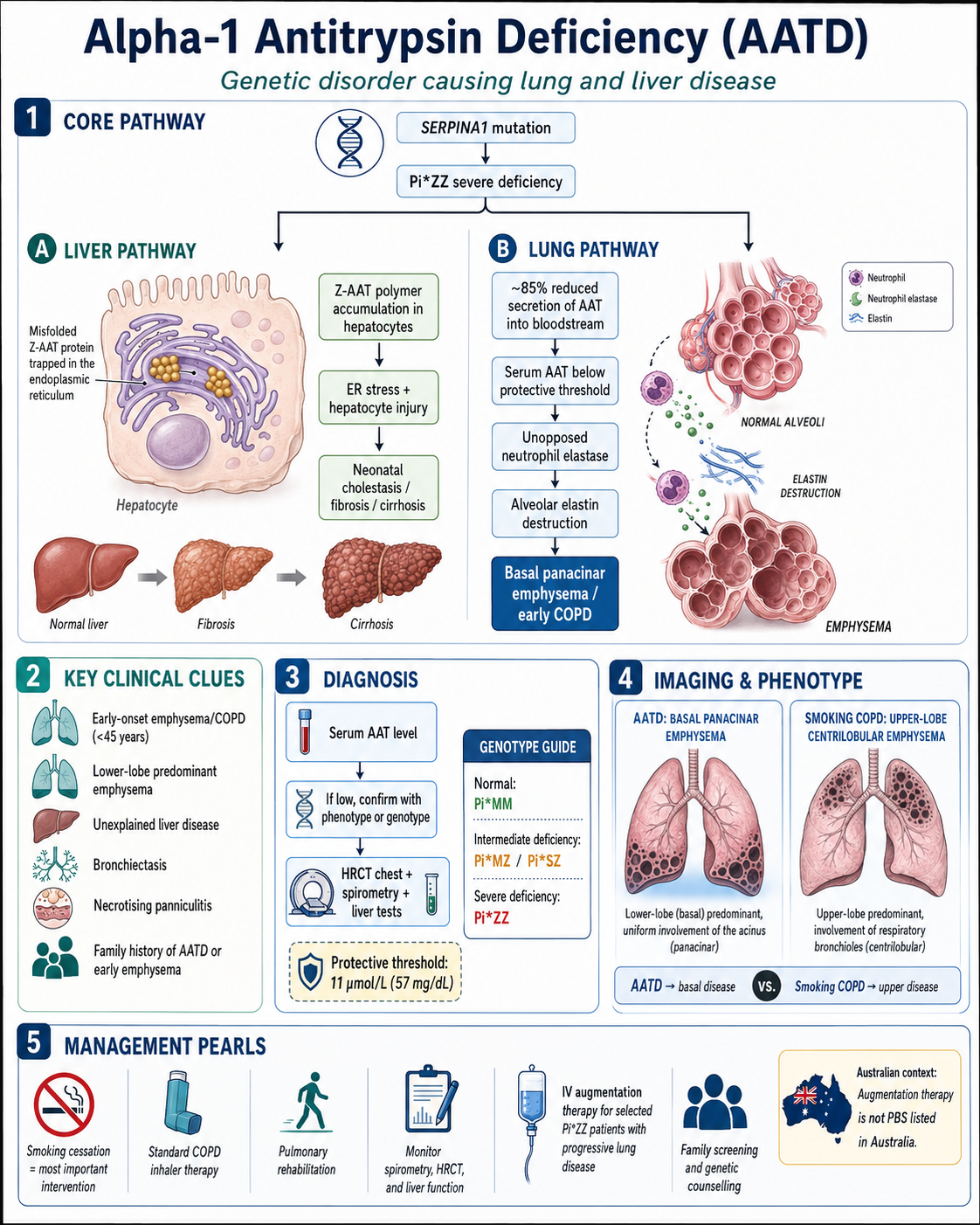
- The most clinically significant deficiency genotype is PI*ZZ, with serum AAT levels <11 µmol/L (~57 mg/dL), conferring a 50–70-fold increased risk of COPD if the individual smokes.
- Screen with serum AAT level in all adults with early-onset COPD (age <45 years), unexplained liver disease, necrotising panniculitis, or a family history of AATD or early emphysema.
- Confirmatory testing requires phenotyping (isoelectric focusing) or genotyping (SERPINA1 gene analysis) — available through Australian pathology services.
- Smoking cessation is the single most important intervention; passive smoke exposure must also be eliminated.
- Standard COPD pharmacotherapy (inhaled bronchodilators ± inhaled corticosteroids) applies as per COPD-X guidelines, but inhaled steroids are not specifically indicated for AATD alone.
- IV alpha-1 antitrypsin augmentation therapy (pooled human AAT) is indicated for PI*ZZ adults with moderate airflow obstruction (FEV₁ 35–60% predicted) who have ceased smoking and have evidence of progressive lung disease.
- Augmentation therapy is NOT currently listed on the PBS in Australia; access is via compassionate use programs, hospital-based procurement, or clinical trials.
- All first-degree relatives of an affected individual should be offered genetic counselling and testing; cascade screening is the most efficient strategy.
- Neonates and children with PI*ZZ may develop cholestatic liver disease; liver function should be monitored in childhood and throughout life.
- Pregnancy in AATD requires multidisciplinary input; augmentation therapy safety in pregnancy is not well established and decisions should be individualised.
- Aboriginal and Torres Strait Islander populations have lower prevalence of classical European AATD alleles but may be underdiagnosed due to reduced access to screening and specialist services in remote areas.
Introduction & Australian Epidemiology
Alpha-1 antitrypsin deficiency (AATD) is one of the most common inherited disorders worldwide and the most common genetic cause of emphysema. It is caused by mutations in the SERPINA1 gene on chromosome 14q32.1, which encodes the serine protease inhibitor alpha-1 antitrypsin (AAT). AAT is primarily synthesised in hepatocytes and functions to protect the lungs from neutrophil elastase-mediated tissue destruction.
Over 150 allelic variants of SERPINA1 have been described, classified by their effect on serum AAT concentration and function: normal (M), deficient (S, Z, and others), null (no protein produced), and dysfunctional (abnormal protein function). The Pi (protease inhibitor) nomenclature uses isoelectric focusing patterns. The normal homozygous genotype is PI*MM, present in approximately 95% of European-descent populations.
In Australia, AATD remains significantly underdiagnosed. It is estimated that fewer than 10% of affected individuals have been identified. Key Australian epidemiological data include:
- The PI*ZZ genotype prevalence is approximately 1 in 1,600–2,500 Australians of European descent.
- The PI*MS carrier frequency is approximately 5–7% and PI*MZ approximately 2–3%.
- The Australian Alpha-1 Antitrypsin Deficiency Registry (hosted by the Thoracic Society of Australia and New Zealand) has enrolled several hundred patients but represents a fraction of estimated cases.
- AATD accounts for an estimated 1–3% of all diagnosed COPD cases in Australia, though true prevalence may be higher given screening gaps.
- Liver disease due to AATD is an important cause of neonatal cholestasis and may present as cryptogenic cirrhosis in adults.
Diagnosis & Screening
Accurate diagnosis of AATD requires a systematic approach: initial serum AAT measurement, followed by phenotyping or genotyping for confirmation, and assessment of end-organ involvement. Screening for AATD should be considered in all patients meeting established criteria.
Screening Indications
The following groups should be screened for AATD, consistent with recommendations from the Thoracic Society of Australia and New Zealand (TSANZ), American Thoracic Society (ATS), and European Respiratory Society (ERS):
- All adults with COPD, regardless of age or smoking history (once-in-lifetime measurement recommended).
- Adults with early-onset emphysema (age <45 years), particularly if lower-lobe predominant on CT.
- Adults with unexplained liver disease or cryptogenic cirrhosis.
- Neonates or infants with prolonged cholestatic jaundice.
- Individuals with necrotising panniculitis.
- First-degree relatives of known AATD patients (cascade screening).
- Adults with bronchiectasis of unknown aetiology.
- Adults with C-ANCA-positive vasculitis (granulomatosis with polyangiitis).
- Population screening is not currently recommended in Australia, but neonatal screening pilot programs have been conducted internationally.
Serum AAT Level
Serum AAT is measured by nephelometry or turbidimetry, widely available through Australian pathology providers (Medicare-rebatable).
| Serum AAT Level | Interpretation | Likely Genotype |
|---|---|---|
| ≥1.3 g/L (≥20 µmol/L) | Normal | PI*MM, PI*MS, PI*SS |
| 0.5–1.2 g/L (9–19 µmol/L) | Intermediate deficiency | PI*MZ, PI*M-null, PI*SZ |
| <0.5 g/L (<11 µmol/L) | Severe deficiency (protease/anti-protease imbalance threshold) | PI*ZZ, PI*Z-null, PI*Null-Null |
Phenotyping and Genotyping
If the serum AAT level is below normal, confirmatory testing is essential:
- Phenotyping — Isoelectric focusing (IEF) of serum to determine the Pi protein variant. Gold standard for identifying S and Z alleles. Available at major Australian pathology laboratories.
- Genotyping — Targeted mutation analysis (PCR-based) for the most common deficiency alleles (S and Z). Does not detect rare or novel variants. Available through specialised genetics laboratories.
- Full gene sequencing — SERPINA1 sequencing when phenotyping and targeted genotyping are discordant or when rare/null alleles are suspected. Refer to clinical genetics service.
Imaging
High-resolution CT (HRCT) chest is recommended at diagnosis to characterise emphysema pattern:
- Characteristic pattern: Basal-predominant, panacinar emphysema — this distribution is relatively specific for AATD and helps differentiate from smoking-related centrilobular emphysema (which is typically upper-zone predominant).
- CT densitometry (quantitative assessment of lung density) may be used as a surrogate biomarker for disease progression and is an endpoint in augmentation therapy trials.
Pathophysiology
Understanding AATD pathophysiology is essential for rational therapeutic decisions. The disease involves two distinct pathological mechanisms — pulmonary and hepatic.
Pulmonary Pathology
- AAT is the primary inhibitor of neutrophil elastase in the lower respiratory tract. When AAT levels fall below the protective threshold of approximately 11 µmol/L (57 mg/dL), an imbalance between protease and anti-protease activity occurs.
- Unopposed neutrophil elastase degrades elastin and other structural proteins in the alveolar walls, leading to progressive alveolar destruction and emphysema.
- Smoking, occupational dust exposure, and airway inflammation amplify neutrophilic inflammation, accelerating lung damage through oxidative inactivation of remaining AAT and increased elastase release.
- The Z protein (Glu342Lys substitution) causes AAT to polymerise within hepatocyte endoplasmic reticulum, reducing secretion by ~85% and generating hepatotoxic intracellular polymers.
Hepatic Pathology
- Accumulation of Z-AAT polymers in hepatocytes triggers endoplasmic reticulum stress, autophagy activation, and hepatocyte injury.
- This may manifest as neonatal cholestasis (in ~10% of PI*ZZ neonates), childhood liver fibrosis, or adult cryptogenic cirrhosis.
- Liver disease is the second most common cause of death in AATD patients and the most common cause of death in children with PI*ZZ.
Other Organ Involvement
- Panniculitis: Rare manifestation; painful, recurrent skin nodules, typically on the trunk and extremities. Due to unopposed elastase activity in subcutaneous tissue.
- Vasculitis: Association with C-ANCA-positive granulomatosis with polyangiitis (GPA), though causality is debated.
- Renal disease: IgA nephropathy and membranoproliferative glomerulonephritis have been reported in AATD cohorts.
Clinical Presentation & Diagnostic Criteria
Pulmonary Presentation
Lung disease is the most common clinical manifestation of AATD, typically presenting in the 3rd–5th decades of life:
- Progressive exertional dyspnoea, initially on exertion, eventually at rest.
- Chronic productive cough with mucoid or mucopurulent sputum.
- Recurrent lower respiratory tract infections, particularly bronchiectasis-associated infections.
- Wheezing — often misdiagnosed as asthma, leading to delayed AATD diagnosis.
- Symptom onset earlier than typical smoking-related COPD (often age 35–45 vs 55+).
- Predominantly lower-lobe emphysema on CT (contrasts with upper-lobe predominant in smoking COPD).
Hepatic Presentation
- Neonates: Prolonged conjugated hyperbilirubinaemia in the first weeks of life; ~10% of PI*ZZ neonates are affected. Most resolve spontaneously.
- Children: Hepatomegaly, elevated transaminases; some progress to fibrosis.
- Adults: May present as cryptogenic cirrhosis, portal hypertension, or hepatocellular carcinoma. Should be considered in any adult with unexplained liver disease.
Other Presentations
- Necrotising panniculitis: Painful, recurrent, erythematous subcutaneous nodules that may ulcerate; responds to dapsone or augmentation therapy.
- Granulomatosis with polyangiitis: C-ANCA-positive vasculitis — screen for AATD.
Management
Management of AATD encompasses general COPD strategies (with some AATD-specific modifications), monitoring for disease progression, and consideration of augmentation therapy. All management should follow a multidisciplinary approach with respiratory specialist involvement.
Smoking Cessation
- Offer pharmacotherapy: varenicline (Champix®) — PBS Authority Required; or nicotine replacement therapy (NRT patches, gum, lozenge) — available OTC; or bupropion (Zyban®) — PBS Restricted Benefit.
- Refer to Quitline (13 7848) for telephone counselling.
- Avoid passive smoke exposure — advise household members and social contacts.
- Cannabis smoking should also be addressed, as it contributes to airway inflammation.
Bronchodilator Therapy
Standard COPD pharmacotherapy applies, aligned with COPD-X and GOLD guidelines:
Pulmonary Rehabilitation
- All symptomatic AATD patients (mMRC ≥2 or CAT ≥10) should be referred for pulmonary rehabilitation.
- Programs are available through public hospitals, community health centres, and some private providers across Australia.
- Improves exercise capacity, dyspnoea, and quality of life. Referred at 12-monthly intervals.
Exacerbation Management
- Treat acute exacerbations per standard COPD protocols: short-acting bronchodilators, systemic corticosteroids (prednisolone 40–50 mg PO daily for 5 days), and antibiotics if purulent sputum.
- Consider sputum culture to guide antibiotic choice, especially if bronchiectasis is present.
- First-line antibiotics: amoxicillin 500 mg PO TDS for 5–7 days, or doxycycline 200 mg PO day 1 then 100 mg daily for 5 days.
- If Pseudomonas aeruginosa is isolated (common in AATD bronchiectasis): ciprofloxacin 750 mg PO BD for 7–10 days or refer for IV therapy.
Monitoring
- Spirometry: Every 6–12 months; track FEV₁ decline. Normal decline in AATD is ~50–150 mL/year (vs ~30 mL/year in non-deficient individuals).
- HRCT chest: At diagnosis and every 2–5 years to monitor emphysema progression (CT densitometry if available).
- Liver function tests: Baseline and annually; more frequently if abnormal.
- Serum AAT level: Confirm level after diagnosis; if on augmentation therapy, check trough level before infusion.
- 6-minute walk test: At baseline and annually in moderate-severe disease.
- Quality of life: CAT score and/or SGRQ at each clinic visit.
Augmentation Therapy
Intravenous alpha-1 antitrypsin augmentation therapy involves regular infusions of pooled human plasma-derived AAT to restore anti-elastase protection in the lungs. It is the only disease-specific therapy currently available for AATD-related emphysema.
Mechanism
- Pooled human AAT (derived from donor plasma) is administered intravenously to raise serum AAT levels above the protective threshold of 11 µmol/L.
- Weekly infusions maintain consistent trough levels, providing sustained anti-neutrophil elastase activity in the alveolar epithelial lining fluid.
- The anti-inflammatory and immunomodulatory properties of AAT may provide additional benefit beyond elastase inhibition.
Eligibility Criteria
- Confirmed PI*ZZ, PI*Z-null, or PI*null-null genotype (or equivalent severe deficiency).
- Serum AAT level <11 µmol/L (57 mg/dL).
- Evidence of lung disease: FEV₁ 35–60% predicted (moderate airflow obstruction).
- Demonstrated rate of FEV₁ decline or CT densitometry decline (progressive disease).
- Patient has ceased smoking (minimum 6 months abstinence; never-smokers also eligible).
- Receiving optimal standard COPD management.
Augmentation therapy is not indicated for:
- PI*MZ, PI*MS, or PI*SZ heterozygotes (these genotypes do not have severe deficiency).
- Patients with FEV₁ >80% predicted (mild disease — insufficient evidence of benefit).
- Patients with FEV₁ <20% predicted (very severe disease — limited evidence of benefit; consider transplant instead).
- Active smokers (augmentation therapy is ineffective while smoking continues).
- Liver-predominant AATD without significant lung disease.
Available Products
Cost Considerations
- Hospital procurement through Special Access Scheme (SAS) or Authorised Prescriber pathway via the TGA.
- Some state health departments provide limited funding for confirmed severe cases with documented progression.
- The Alpha-1 Foundation Australia and patient advocacy groups may assist with navigating access.
- Home infusion programs can reduce healthcare system burden and improve patient quality of life where available.
Monitoring Efficacy of Augmentation Therapy
- Trough serum AAT level: Measure before the next infusion; target ≥11 µmol/L. Check at 1 month, then every 3–6 months.
- Spirometry: Every 6 months; compare rate of FEV₁ decline to pre-treatment trajectory. A reduction in the rate of decline suggests efficacy.
- CT densitometry: Annually if available; validated as a sensitive biomarker of emphysema progression (RAPID study).
- Exacerbation frequency: Document and compare to pre-treatment baseline.
- Quality of life: SGRQ or CAT score at 3-monthly intervals initially, then 6-monthly.
Family Screening & Genetic Counselling
Cascade screening of first-degree relatives is the most efficient strategy for identifying undiagnosed AATD cases and is strongly recommended by TSANZ, ATS, and ERS. Early identification enables risk modification (smoking avoidance), surveillance, and timely intervention.
Genetic Counselling
- Genetic counselling should be offered to all individuals diagnosed with AATD and their first-degree relatives.
- Counselling should cover: autosomal codominant inheritance pattern, implications of specific genotype (PI*ZZ vs PI*MZ), risk to offspring, pulmonary and hepatic implications, and the importance of smoking avoidance.
- Psychosocial support should be offered, including referral to the Alpha-1 Foundation Australia and relevant support groups.
- Genetic discrimination concerns: advise patients about Australian privacy protections and the Genetic Discrimination Project. The Disability Discrimination Act 1992 (Cth) provides some protection.
Testing Relatives
- First-degree relatives (parents, siblings, children) of a confirmed AATD patient should be offered testing.
- Start with serum AAT level — if abnormal, proceed to phenotyping/genotyping.
- Alternatively, direct genotyping for the known family allele may be appropriate if the proband's genotype is confirmed.
- Testing children: serum AAT level can be measured at any age. If deficiency is confirmed, monitor liver function, counsel regarding smoking avoidance, and plan ongoing respiratory surveillance.
- At-risk relatives who are PI*MZ carriers should be counselled about their near-normal AAT levels but heightened risk if they smoke and the importance of avoiding occupational exposures.
Liver Disease Screening
- All PI*ZZ individuals should have baseline liver function tests (ALT, AST, GGT, bilirubin, albumin, INR) at diagnosis.
- PI*ZZ neonates with prolonged jaundice should be evaluated by a paediatric hepatologist.
- Annual LFTs for all PI*ZZ individuals throughout life.
- Transient elastography (FibroScan®) or ultrasound-based elastography may be used for non-invasive liver fibrosis assessment if LFTs are abnormal.
- Hepatocellular carcinoma surveillance (6-monthly ultrasound and AFP) should be considered in PI*ZZ patients with established cirrhosis.
Pregnancy Considerations
- AATD does not directly impair fertility; however, severe COPD may complicate pregnancy and require obstetric-respiratory multidisciplinary management.
- Augmentation therapy in pregnancy: limited safety data. Risk-benefit analysis should be individualised. If augmentation is deemed necessary (severe, progressive lung disease), it may be continued with informed consent and close monitoring.
- If both parents are PI*MZ, there is a 25% chance of each child being PI*ZZ. Offer prenatal or pre-implantation genetic counselling.
- PI*ZZ mothers can breastfeed; AAT is present in breast milk but levels are low and breastfeeding does not significantly affect infant serum AAT.
- Standard vaccines (pertussis, influenza, COVID-19) should be administered per Australian Immunisation Handbook recommendations.
Special Populations
Paediatrics
- PI*ZZ neonates: ~10% develop cholestatic jaundice; most resolve spontaneously by 6 months. Monitor LFTs closely.
- Refer persistent neonatal cholestasis to paediatric hepatology.
- Childhood liver fibrosis may occur; annual LFTs and clinical assessment recommended.
- Smoking avoidance counselling should begin in adolescence — primary prevention is critical.
- Spirometry from age 6–8 years if clinically indicated.
- Augmentation therapy is not indicated in children.
Pregnancy
- Mild-moderate AATD generally tolerates pregnancy well with close respiratory monitoring.
- Severe AATD (FEV₁ <50%): high-risk obstetric input required; risk of respiratory decompensation.
- Augmentation therapy: limited safety data; risk-benefit discussion essential.
- Avoid teratogenic medications; review bronchodilators for pregnancy safety.
- Genetic counselling regarding offspring genotype.
Elderly
- Comorbidities common: osteoporosis (steroid use, smoking history), cardiovascular disease, anxiety/depression.
- Osteoporosis screening (DEXA) should be performed given corticosteroid exposure history.
- Polypharmacy review — avoid anticholinergic burden with LAMA if cognitive impairment.
- Frailty assessment and falls risk if supplemental oxygen required.
- Advance care planning should be discussed.
Renal Impairment
- IgA nephropathy and membranoproliferative glomerulonephritis are recognised associations — screen with urinalysis and renal function.
- Augmentation therapy does not require renal dose adjustment.
- Avoid nephrotoxic drugs where possible; dose-adjust antibiotics if eGFR <30 mL/min.
Hepatic Impairment
- Liver disease is intrinsic to AATD — does not improve with augmentation therapy (which replaces AAT in blood, not hepatocytes).
- Avoid hepatotoxins (alcohol, unnecessary hepatotoxic medications).
- Liver transplant is the definitive treatment for end-stage liver disease due to AATD and cures the metabolic defect.
- Combined lung-liver transplant may be considered in patients with severe pulmonary and hepatic disease.
Immunocompromised
- AATD patients are not inherently immunocompromised but may be on immunosuppressive therapy for comorbid autoimmune conditions.
- Augmentation therapy products are derived from pooled human plasma — theoretical risk of pathogen transmission, though modern manufacturing minimises this risk.
- Live vaccines may be contraindicated if on immunosuppressants — check Australian Immunisation Handbook.
- Pneumococcal and influenza vaccination strongly recommended for all AATD patients.
AATD in Aboriginal and Torres Strait Islander populations requires specific consideration. The classical European PI*S and PI*Z alleles are uncommon in Indigenous Australian populations; however, other genetic variants affecting AAT may exist and screening gaps remain significant.
📚 References
- 1. American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900.
- 2. Stockley RA, Miravitlles M, Vogelmeier C, et al. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149.
- 3. Chapman KR, Burdon JGW, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–368.
- 4. Alpha One Foundation. Alpha-1 antitrypsin deficiency: a guide for patients and families. 7th ed. Miami, FL: Alpha One Foundation; 2022.
- 5. Greve S, Goggs R, Knight D, et al. Alpha-1 antitrypsin deficiency in Australia: a review. Respirology. 2020;25(6):596–604.
- 6. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: 2024 report. Fontana, WI: GOLD; 2024.
- 7. Thoracic Society of Australia and New Zealand (TSANZ). The COPD-X Plan: Australian and New Zealand guidelines for the management of chronic obstructive pulmonary disease. Version 2.69. Sydney: TSANZ; 2024.
- 8. Australian Institute of Health and Welfare (AIHW). Chronic obstructive pulmonary disease (COPD). Cat. no. ACM 35. Canberra: AIHW; 2023.
- 9. McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017;5(1):51–60.
- 10. Strnad P, McElvaney NG, Lomas DA. Alpha1-antitrypsin deficiency. N Engl J Med. 2020;382(15):1443–1455.
- 11. Dirksen A, Piitulainen E, Deng C, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha-1 antitrypsin deficiency. Eur Respir J. 2009;33(6):1345–1353.
- 12. Greve SM, Wilson R, Burdon J, et al. Australian experience with augmentation therapy for alpha-1 antitrypsin deficiency. Intern Med J. 2018;48(12):1472–1477.
- 13. RACGP. Handbook of non-drug interventions (HANDI): Smoking cessation. Melbourne: Royal Australian College of General Practitioners; 2023.
- 14. Teckman J, Jain A. Advances in alpha-1 antitrypsin deficiency liver disease. Curr Gastroenterol Rep. 2014;16(4):367.
- 15. Edgar RG, Patel M, Bayliss S, et al. Treatment of lung disease in alpha-1 antitrypsin deficiency: a systematic review. Int J Chron Obstruct Pulmon Dis. 2017;12:1295–1308.