📋 Key Information Summary
- Cystic fibrosis (CF) is the most common life-limiting autosomal recessive condition in Australia, affecting approximately 1 in 2,500–3,000 Caucasian births, with over 3,500 Australians currently living with CF.
- Newborn bloodspot screening (NBS) is universal across all Australian states and territories since 2018; an immunoreactive trypsinogen (IRT)–DNA or IRT–IRT–DNA protocol is used, followed by confirmatory sweat chloride testing (≥60 mmol/L diagnostic).
- CFTR genotype determines eligibility for CFTR modulator therapy; over 2,000 mutations identified; F508del is carried by ~1 in 25 Australians and accounts for ~70% of CF alleles.
- Elexacaftor/tezacaftor/ivacaftor (Trikafta®) is a transformative therapy for patients with at least one F508del allele (~85–90% eligible), improving FEV₁ by 10–14% predicted and dramatically reducing pulmonary exacerbations. It was PBS-listed in Australia from November 2022 as Authority Required.
- Pancreatic insufficiency occurs in ~85% of infants with CF; pancreatic enzyme replacement therapy (PERT) with lipase dosing of 500–4,000 units/kg/day is mandatory, titrated by symptoms and faecal elastase.
- Chronic Pseudomonas aeruginosa infection remains the leading cause of morbidity; early eradication with inhaled tobramycin (TOBI®) or colistin plus oral ciprofloxacin is standard; chronic suppressive therapy uses alternating inhaled antibiotics.
- CF-related diabetes (CFRD) affects up to 50% of adults with CF; annual oral glucose tolerance testing (OGTT) should commence from age 10 years; insulin is the primary treatment, with early referral to an endocrinologist.
- All people with CF should receive regular airway clearance techniques (ACTs) guided by a physiotherapist, with hypertonic saline (7%) and dornase alfa (Pulmozyme®) as key mucolytic therapies.
- Burkholderia cepacia complex and non-tuberculous mycobacteria (NTM) require specialist infectious diseases input; B. cenocepacia (genomovar III) is a relative contraindication to lung transplant at many Australian centres.
- Lung transplantation should be discussed proactively; referral when FEV₁ falls below 50% predicted or with rapid decline, recurrent haemoptysis, or refractory pneumothorax. Median post-transplant survival in Australia exceeds 8 years.
- Aboriginal and Torres Strait Islander Australians with CF face delayed diagnosis, reduced access to CFTR modulators, and higher rates of chronic Pseudomonas infection; culturally safe, multidisciplinary care with remote-health integration is essential.
- Multidisciplinary CF care (respiratory physician, dietitian, physiotherapist, CF nurse specialist, psychologist, social worker, pharmacist) at an accredited CF centre is associated with superior outcomes and is the standard of care in Australia.
Introduction & Australian Epidemiology
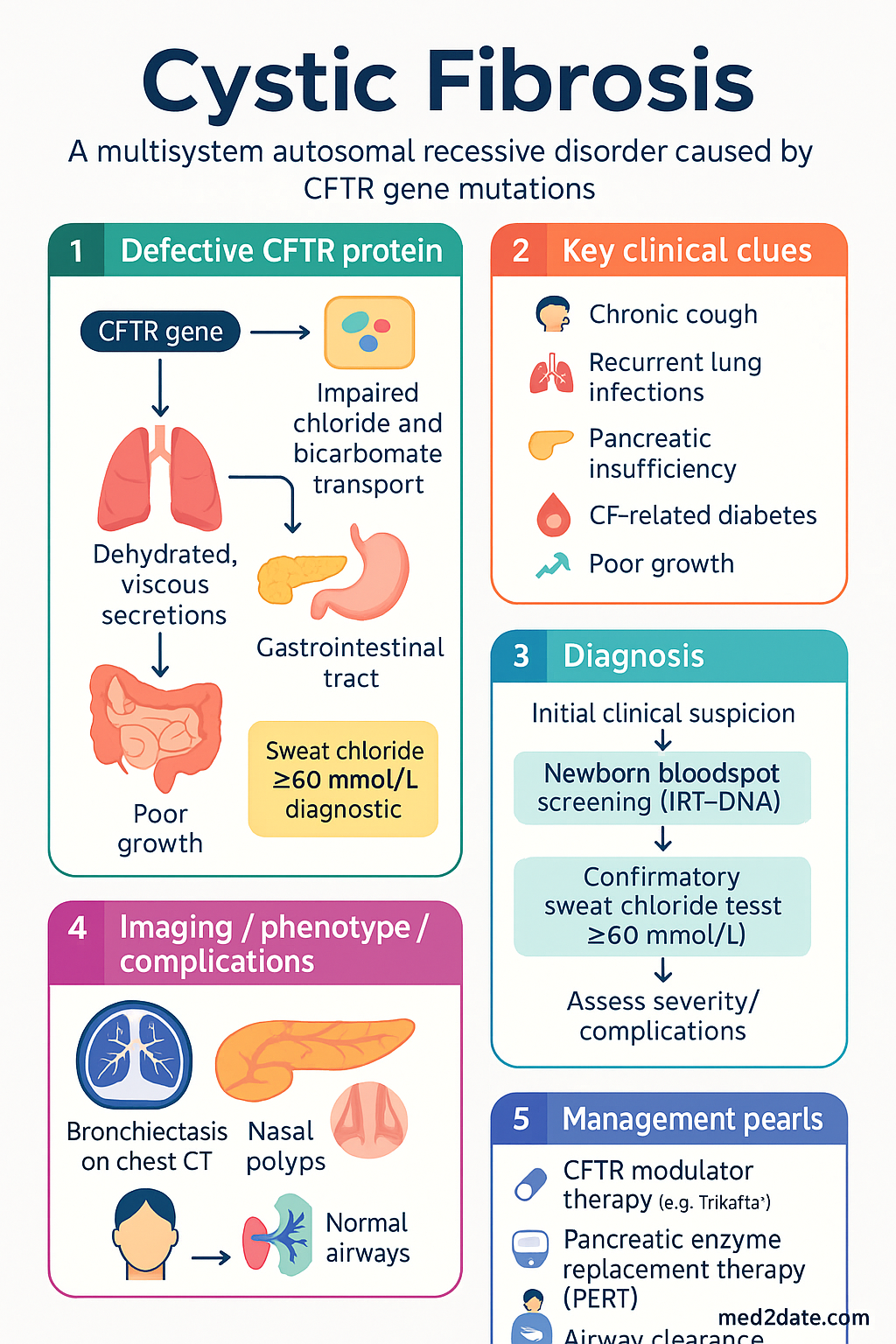
Cystic fibrosis (CF) is a multisystem autosomal recessive disorder caused by pathogenic variants in the CFTR (cystic fibrosis transmembrane conductance regulator) gene located on chromosome 7q31.2. The defective CFTR protein impairs epithelial chloride and bicarbonate transport, leading to dehydrated, viscous secretions in the lungs, pancreas, hepatobiliary system, gastrointestinal tract, and reproductive tract.
CF is the most common life-limiting genetic condition among Australians of European descent. The Australian Cystic Fibrosis Data Registry (ACFDR), maintained by Cystic Fibrosis Australia, records approximately 3,500 people living with CF nationally. Carrier frequency is approximately 1 in 25 in populations of Northern European ancestry.
| Epidemiological Parameter | Australian Data |
|---|---|
| Incidence (Caucasian) | 1 in 2,500–3,000 live births |
| Carrier frequency | ~1 in 25 (European ancestry) |
| Total prevalence (people living with CF) | ~3,500 (ACFDR 2022 report) |
| Median predicted survival (born 2022) | ~55–60 years (improving with modulators) |
| New diagnoses per year | ~100–120 (predominantly via NBS) |
| Indigenous Australians | Lower reported incidence but likely under-diagnosis; higher rates of late presentation |
Since the introduction of universal newborn screening across all Australian jurisdictions (completed 2018), the majority of CF diagnoses are now made in the first 4–6 weeks of life, enabling early intervention and improved long-term outcomes. The establishment of accredited CF centres at major tertiary hospitals in each state and territory ensures multidisciplinary care delivery aligned with Cystic Fibrosis Australia standards.
Diagnosis & Genetics
Newborn Screening (NBS)
Universal newborn bloodspot screening is performed in all Australian states and territories. Most jurisdictions use an IRT–DNA or IRT–IRT–DNA algorithm:
- Step 1: Immunoreactive trypsinogen (IRT) measured on day 2–4 heel-prick bloodspot; elevated IRT (typically >99th percentile) triggers further testing.
- Step 2: CFTR mutation panel analysis (varies by state; typically 20–50 common mutations including F508del, G551D, G542X, etc.). Some states use a second IRT at 2–4 weeks before DNA analysis.
- Step 3: Referral to a paediatric CF centre for confirmatory sweat chloride testing within the first 4–8 weeks of life.
Sweat Chloride Testing
The sweat chloride test (pilocarpine iontophoresis) remains the gold standard for CF diagnosis. It must be performed in an accredited laboratory following the Gibson–Cooke or Macroduct methodology (CLSI C34-A3).
| Sweat Chloride (mmol/L) | Interpretation |
|---|---|
| < 30 | Normal — CF unlikely |
| 30–59 | Intermediate — consider CFTR-related disorder, repeat testing, or genetic analysis |
| ≥ 60 | Positive — consistent with CF (in appropriate clinical context) |
CFTR Mutation Analysis
Comprehensive CFTR genotyping is recommended for all individuals with a positive NBS, intermediate sweat chloride, or clinical features suggestive of CF. Next-generation sequencing (NGS) panels covering all 27 exons and splice sites are available at Australian genetic pathology laboratories (MBS item 73291 and related).
CFTR mutations are classified into six functional classes:
| Class | Mechanism | Examples | CFTR Modulator Response |
|---|---|---|---|
| I | No functional protein (nonsense, frameshift) | G542X, W1282X, R553X | Limited — no response to current modulators (pipeline: gene therapy, read-through agents) |
| II | Processing/trafficking defect | F508del (most common) | Excellent — elexacaftor/tezacaftor/ivacaftor (Trikafta®) |
| III | Gating defect (reduced open probability) | G551D, G178R | Good — ivacaftor (Kalydeco®) monotherapy |
| IV | Conductance defect (reduced ion flow) | R117H, R347P | Partial — ivacaftor may help (R117H) |
| V | Reduced protein quantity (splicing) | 3849+10kbC→T, A455E | Variable — may respond to potentiators |
| VI | Increased protein turnover (instability) | Rescued F508del | May respond to stabilising correctors |
Pancreatic Status Assessment
Faecal elastase-1 (FE-1) is the preferred non-invasive test for pancreatic exocrine function:
- > 200 µg/g: Pancreatic sufficient (PS) — ~15% of CF patients
- 100–200 µg/g: Mildly insufficient — monitor closely
- < 100 µg/g: Pancreatic insufficient (PI) — requires PERT (~85% of CF patients)
CFTR Modulator Therapy
CFTR modulators target the underlying protein defect rather than symptoms alone. They are classified as potentiators (improve gating of CFTR at the cell surface), correctors (improve folding and trafficking of CFTR to the cell surface), and amplifiers (increase CFTR mRNA). The availability of elexacaftor/tezacaftor/ivacaftor on the Australian PBS has been the single greatest advance in CF therapeutics.
Genotype–Modulator Matching Summary
| CFTR Genotype | Recommended Modulator | Australian PBS |
|---|---|---|
| ≥1 F508del allele (most patients) | Elexacaftor/tezacaftor/ivacaftor (Trikafta®) | PBS Authority Required |
| Gating mutations (G551D, etc.) without F508del | Ivacaftor (Kalydeco®) | PBS Authority Required |
| R117H (residual function) | Ivacaftor (Kalydeco®) | PBS Authority Required |
| Homozygous F508del (if Trikafta contraindicated) | Lumacaftor/ivacaftor (Orkambi®) or tezacaftor/ivacaftor (Symdeko®) | PBS Authority Required |
| Two minimal function mutations (no F508del, no gating) | No current modulator; gene therapy trials; refer to CF specialist | N/A |
Pulmonary Management
Pulmonary disease accounts for the majority of CF morbidity and mortality. The objectives of pulmonary management are to clear airway secretions, prevent and treat lung infections, reduce inflammation, and maintain lung function. All pulmonary management should be guided by an accredited CF physiotherapist and respiratory physician.
Airway Clearance Techniques (ACTs)
Daily airway clearance is the cornerstone of CF lung care. The specific regimen should be individualised by a CF physiotherapist based on age, disease severity, patient preference, and sputum characteristics.
Inhaled Mucolytic Therapies
Inhaled Antibiotics — Maintenance
Inhaled Bronchodilators
Short-acting β₂-agonists (salbutamol 200–400 µg MDI via spacer or 2.5–5 mg nebulised) should be administered before airway clearance and hypertonic saline. Long-acting bronchodilators (salmeterol, formoterol) may be added for patients with documented bronchospasm or reversible obstruction on spirometry.
Inhaled Corticosteroids
Inhaled corticosteroids (ICS) are not routinely recommended for all CF patients. They should be considered only when there is a clear comorbid asthma or CF-associated allergic bronchopulmonary aspergillosis (ABPA). Unnecessary ICS use may increase the risk of NTM infection and adrenal suppression. If used, the lowest effective dose should be prescribed with regular reassessment.
Routine Pulmonary Monitoring
| Investigation | Frequency | Purpose |
|---|---|---|
| Spirometry (FEV₁, FVC) | Every clinic visit (minimum quarterly) | Track lung function trajectory; detect early decline |
| Sputum culture / cough swab | Every clinic visit (minimum quarterly) | Identify pathogens; guide antibiotic therapy |
| CT chest (HRCT) | Every 2–4 years or as clinically indicated | Structural lung disease assessment; bronchiectasis severity |
| Chest X-ray | Annually or with acute exacerbations | Screening for complications (pneumothorax, etc.) |
| Exercise testing (6MWT / CPET) | Annually | Functional capacity; prognostic indicator |
| Pulse oximetry | Every visit and during acute illness | Detect hypoxaemia |
Infection Management
Lung infection is the primary driver of morbidity and mortality in CF. The microbiology of the CF airway evolves over time, with characteristic pathogens at different life stages. Eradication of initial infection and aggressive treatment of pulmonary exacerbations are fundamental to preserving lung function.
Typical Microbial Progression
Pseudomonas aeruginosa Eradication Protocol
Standard Australian eradication regimen (eTG-aligned):
- Oral ciprofloxacin 20–30 mg/kg/day (max 1.5 g/day) in two divided doses for 2–3 weeks PLUS
- Inhaled tobramycin 300 mg nebulised every 12 hours for 4 weeks (TOBI®) OR
- Inhaled colistimethate 1–2 million units every 12 hours for 4 weeks as alternative to tobramycin
- Follow-up sputum culture at 1 month after completion; if still positive, consider IV anti-pseudomonal therapy (see below)
Chronic Pseudomonas — Suppressive Therapy
Patients with chronic P. aeruginosa infection (defined as ≥3 positive cultures in the preceding 12 months, or persistently positive for >6 months) should receive continuous alternating inhaled antibiotic therapy:
- Month 1: Inhaled tobramycin 300 mg every 12 hours
- Month 2: Inhaled colistimethate 1–2 million units every 12 hours OR inhaled aztreonam 75 mg TDS
- Alternate monthly; continue indefinitely unless eradicated (rare in chronic infection)
Pulmonary Exacerbations — IV Antibiotic Therapy
Indications for IV antibiotics include declining FEV₁ (>10% from baseline), increased sputum volume/purulence, systemic symptoms (fever, weight loss, reduced appetite), or failure of oral/inhaled therapy. IV therapy is typically administered for 14 days, often as an inpatient or via Hospital in the Home (HITH).
| Pathogen | First-Line IV Regimen | Second-Line / Notes |
|---|---|---|
| P. aeruginosa | Piperacillin–tazobactam 4.5 g IV every 6 hours + tobramycin 5–7 mg/kg IV once daily (extended-interval) — or ceftazidime 2 g IV every 8 hours + tobramycin | Meropenem 1–2 g IV every 8 hours if resistant; colistin IV for MDR strains (nephrotoxic — specialist-guided). Monitor tobramycin trough (<1 mg/L for extended-interval). |
| S. aureus (MSSA) | Flucloxacillin 2 g IV every 6 hours (or cefazolin 2 g IV every 8 hours if penicillin allergy non-anaphylaxis) | Duration 10–14 days |
| S. aureus (MRSA) | Vancomycin IV (target trough 15–20 mg/L) ± rifampicin 300–450 mg PO BD | Linezolid 600 mg PO/IV BD for 10–14 days if vancomycin intolerant. Trimethoprim–sulfamethoxazole (TMP-SMX) 160/800 mg PO BD for minor exacerbations. Monitor FBC with linezolid (>2 weeks: thrombocytopenia). |
| B. cepacia complex | Meropenem 1–2 g IV every 8 hours + TMP-SMX 160/800 mg PO BD ± ceftazidime — guided by susceptibilities | Requires specialist ID input. B. cenocepacia (genomovar III) has highest morbidity. No reliable inhaled options. |
| NTM (M. abscessus complex) | Multidrug regimen: amikacin IV + imipenem IV + tigecycline or azithromycin — guided by speciation and susceptibility | Prolonged treatment (≥12 months after sputum conversion). Requires specialist NTM/ID input. Macrolide monotherapy must be avoided (resistance induction). |
Methicillin-Resistant Staphylococcus aureus (MRSA)
CA-MRSA prevalence in Australian CF populations is increasing, particularly in Northern Territory and remote communities. Eradication should be attempted on first isolation:
- Nasal mupirocin 2% ointment BD to each nostril for 5 days + chlorhexidine body washes daily for 5 days
- Consider adding oral TMP-SMX 160/800 mg BD or oral rifampicin 300 mg BD + fusidic acid 500 mg TDS for 2–4 weeks if persistent colonisation
- Chronic MRSA infection: suppressive therapy with TMP-SMX or linezolid (monitor FBC); vancomycin IV for exacerbations
Non-Tuberculous Mycobacteria (NTM)
Mycobacterium abscessus complex is the most clinically significant NTM in CF. It is associated with accelerated lung function decline and can be a relative contraindication to lung transplant at some centres. M. avium complex (MAC) also occurs. Diagnosis requires:
- Positive mycobacterial sputum culture (≥2 positive cultures, or 1 positive with CT findings suggestive of NTM) per ATS/IDSA criteria
- Speciation to species/ subspecies level (e.g., M. abscessus subsp. abscessus vs massiliense) — critical for treatment decisions
- Referral to specialist NTM service (Royal Melbourne Hospital, Westmead Hospital, Royal Adelaide Hospital, Sir Charles Gairdner Hospital have expertise)
Allergic Bronchopulmonary Aspergillosis (ABPA)
ABPA occurs in 2–9% of CF patients and presents with worsening asthma symptoms, fleeting pulmonary infiltrates, elevated total IgE (>500 IU/mL, typically >1,000), positive Aspergillus-specific IgE/IgG, and eosinophilia. Treatment:
- First-line: Oral prednisolone 0.5–1 mg/kg/day (max 40–60 mg) for 1–2 weeks, then taper over 2–3 months
- Steroid-sparing / steroid-refractory: Itraconazole 200 mg PO BD (monitor levels, CYP interactions) or voriconazole 200 mg PO BD
- Biologic therapy: Omalizumab (Xolair®) is increasingly used for refractory ABPA (PBS Authority Required, under specialist supervision)
Multisystem Complications
Pancreatic Exocrine Insufficiency
Approximately 85% of CF patients have pancreatic insufficiency (PI), diagnosed by faecal elastase-1 <100 µg/g. Untreated PI leads to fat malabsorption, steatorrhoea, fat-soluble vitamin deficiency, failure to thrive, and distal intestinal obstruction syndrome (DIOS).
Distal Intestinal Obstruction Syndrome (DIOS)
DIOS results from inspissated faecal material in the ileocaecal region, presenting with crampy abdominal pain, palpable right iliac fossa mass, and constipation. Risk factors include PI, dehydration, immobility, and CFTR modulator initiation (may paradoxically alter stool consistency).
- Mild DIOS: Oral polyethylene glycol (PEG/Movicol®) 1–2 sachets BD; increase PERT dose; encourage fluids
- Moderate–severe DIOS: Gastrografin® (diatrizoate meglumine) 50–100 mL PO/NG diluted 1:1 with water; or balanced PEG-electrolyte solution via NG tube; IV fluids if dehydrated
- Refractory: Surgical consultation — differentiate from meconium ileus equivalent, intussusception, or distal intestinal tumour
Fat-Soluble Vitamins
CF-Related Diabetes (CFRD)
CFRD affects approximately 20% of adolescents and up to 50% of adults with CF. It results from progressive pancreatic fibrosis and insulin deficiency, compounded by CFTR-related β-cell dysfunction. CFRD is associated with accelerated decline in lung function, worse nutritional status, and increased mortality.
| Screening Protocol | Details |
|---|---|
| Age to start | From age 10 years (annually) |
| Test | 2-hour oral glucose tolerance test (OGTT): 75 g glucose load |
| Fasting glucose ≥7.0 mmol/L or 2-hr ≥11.1 mmol/L | CFRD — start insulin |
| Fasting 6.1–6.9 or 2-hr 7.8–11.0 | Impaired glucose tolerance / indeterminate — closer monitoring, consider insulin |
| HbA1c | Not reliable for screening in CF (often normal due to increased red cell turnover and nutritional supplements). Use for monitoring once diagnosed. |
Treatment of CFRD:
- Insulin is the primary therapy — there is no role for sulfonylureas or metformin in CFRD
- Basal–bolus insulin regimen (e.g., insulin glargine once daily + insulin aspart with meals) is the standard approach
- CF patients require higher caloric intake — do NOT restrict carbohydrates; dose insulin to cover nutritional needs
- Continuous glucose monitoring (CGM) is increasingly used (FreeStyle Libre® — PBS-listed for diabetes in some contexts)
- Refer to endocrinologist with CF experience; collaborative CF-endocrine clinics exist at major CF centres
CF-Related Liver Disease
Hepatobiliary disease affects 2–40% of CF patients depending on definition. It ranges from neonatal cholestasis and hepatic steatosis to focal biliary cirrhosis and multilobular cirrhosis.
- Ursodeoxycholic acid (UDCA) 10–20 mg/kg/day in 2–3 divided doses is recommended for patients with evidence of CF-associated hepatopathy (elevated GGT, abnormal ultrasound) — PBS-listed for CF liver disease
- Annual liver function tests (LFTs) and liver ultrasound every 2–3 years from age 3 years
- Monitor for portal hypertension (thrombocytopenia, splenomegaly, varices); upper GI endoscopy if signs of portal hypertension
- CF-related cirrhosis with portal hypertension is an indication for combined lung–liver transplant evaluation
Nutritional Management
A high-calorie, high-fat diet (35–45% of energy from fat for PI patients) with adequate protein is essential. CF dietitians are integral to the multidisciplinary team.
- Caloric targets: 110–200% of estimated average requirement (EAR) for age; individualised by a CF dietitian
- Oral nutritional supplements (e.g., Sustagen®, Ensure Plus®) for those failing to meet targets
- Enteral feeding (nasogastric or gastrostomy) for patients with BMI <25th percentile or failing to thrive despite oral supplementation — common in adolescents and adults with advanced disease
- CFTR modulator therapy often improves appetite and weight; monitor and adjust caloric targets accordingly
Bone Disease
Low bone mineral density (BMD) is common in CF due to chronic inflammation, malabsorption of vitamin D and calcium, delayed puberty, corticosteroid use, and hypogonadism. DEXA scan should be performed from age 8–10 years and repeated every 2–5 years. Calcium (1,000–1,500 mg/day) and vitamin D supplementation (target 25-OH-vitamin D >75 nmol/L) are standard. Bisphosphonates may be considered for adults with T-score ≤ −2.5 or fragility fracture, under specialist guidance.
Lung Transplant Evaluation
Indications for transplant referral:
- FEV₁ <30% predicted (or <50% with rapid decline >20% in 12 months)
- Increasing frequency/severity of pulmonary exacerbations requiring IV antibiotics
- Recurrent massive haemoptysis not amenable to embolisation
- Refractory pneumothorax
- Respiratory failure (hypoxia, hypercapnia)
- CFRD with declining FEV₁
- Deteriorating nutritional status despite maximal support
Contraindications / relative contraindications: Active B. cenocepacia (genomovar III) infection (many Australian centres), active NTM with uncontrolled infection, active malignancy, non-compliance, active substance abuse, severe psychosocial issues. Chronic P. aeruginosa is NOT a contraindication.
Special Populations
Pregnancy
- Preconception planning is essential. Optimise lung function (FEV₁ >50% predicted ideally), nutrition (BMI >22), and glycaemic control before conception.
- CFTR modulators: Elexacaftor/tezacaftor/ivacaftor, lumacaftor/ivacaftor, and tezacaftor/ivacaftor are NOT recommended in pregnancy (teratogenicity data limited; animal studies show some concern). Ivacaftor monotherapy: limited data — discuss risk–benefit with the patient and CF team. Discontinuation of modulator therapy should be carefully weighed against the risk of disease progression.
- Fertility: ~98% of CF males are infertile due to congenital bilateral absence of vas deferens (CBAVD). Assisted reproduction (TESE/IVF) is available. Female fertility may be reduced but is often preserved, especially with adequate nutrition.
- Antenatal monitoring: Joint CF-obstetric clinic; fetal growth surveillance; diabetes management; oxygen saturation monitoring; physiotherapy continuation.
- Delivery: Vaginal delivery preferred unless obstetric indication for caesarean section. Epidural analgesia is safe.
- Breastfeeding: Encouraged; increased caloric requirements (additional 500 kcal/day); most CF medications are compatible with breastfeeding — check with pharmacist.
Paediatrics
- Newborn screening enables diagnosis within 4–6 weeks; early referral to a paediatric CF centre is mandatory.
- PERT should commence at diagnosis for PI infants; dose 2,000–4,000 units lipase per 120 mL of breast milk/formula feed or per meal (titrate by stool consistency and growth).
- Routine immunisations: standard Australian schedule PLUS annual influenza vaccine and pneumococcal vaccine (13vPCV + 23vPPV).
- CFTR modulator therapy: ivacaftor from age ≥1 month (gating mutations); elexacaftor/tezacaftor/ivacaftor from age ≥6 years (PBS-listed); clinical trials for younger ages ongoing.
- Psychosocial support: address school attendance, peer relationships, medication burden, transition to adult services from age 16–18.
- Growth monitoring at every visit; refer to endocrinology if declining height velocity or BMI <25th percentile.
Elderly
- An increasing number of Australians with CF survive into their 50s and 60s, presenting new challenges in geriatric CF care.
- Polypharmacy: increased risk of drug interactions with CFTR modulators (CYP3A). Regular medication review by a CF pharmacist.
- Comorbidities: cardiovascular disease, osteoporosis, renal impairment, malignancy screening (colorectal cancer risk increased in CF — colonoscopy recommended from age 40, every 2–5 years).
- Bone health: DEXA every 2 years; ensure adequate calcium, vitamin D; bisphosphonates as indicated.
- Renal function decline: aminoglycoside toxicity cumulative — limit courses, use extended-interval dosing, monitor creatinine and audiometry.
Renal Impairment
- CF-related kidney disease: nephrotoxicity from aminoglycosides (cumulative exposure), diabetes, and amyloidosis. Prevalence of CKD in CF adults 10–25%.
- Aminoglycoside dosing: extended-interval (once daily) tobramycin (5–7 mg/kg) preferred; therapeutic drug monitoring (TDM) essential — trough <1 mg/L. Use ABW/IBW calculation.
- Annual renal function (eGFR, urine ACR) from age 18 years or after significant aminoglycoside exposure.
- Renal transplant in CF patients with ESRD is feasible and performed at Australian transplant centres; combined lung–kidney transplant may be considered.
Hepatic Impairment
- CF liver disease: monitor LFTs annually; liver ultrasound every 2–3 years from age 3 years.
- UDCA 10–20 mg/kg/day for hepatopathy (PBS-listed for CF liver disease).
- CFTR modulators: lumacaftor/ivacaftor (Orkambi®) contraindicated in severe hepatic impairment (Child–Pugh C). Elexacaftor/tezacaftor/ivacaftor: use with caution in Child–Pugh B; not recommended in Child–Pugh C. Enhanced LFT monitoring required.
- Portal hypertension management: beta-blockers, variceal surveillance endoscopy. Combined lung–liver transplant for decompensated cirrhosis.
Immunocompromised
- CF patients are not classically immunocompromised but have impaired mucociliary clearance and defective innate immunity in the airway, increasing infection susceptibility.
- Post-transplant patients are on immunosuppressive regimens (tacrolimus, mycophenolate, prednisolone) — standard transplant infectious diseases protocols apply.
- Annual influenza vaccine (inactivated) for all CF patients and household contacts; COVID-19 vaccination per ATAGI recommendations; pneumococcal vaccination (13vPCV + 23vPPV).
- CMV and EBV surveillance post-transplant; Pneumocystis jirovecii prophylaxis with TMP-SMX post-transplant.
Aboriginal and Torres Strait Islander Health
CF is less commonly diagnosed in Aboriginal and Torres Strait Islander populations, but when identified, it is associated with delayed diagnosis, reduced access to CFTR modulator therapy, higher rates of chronic Pseudomonas infection, and poorer health outcomes. Culturally safe, multidisciplinary care is essential to close the gap in CF outcomes.
📚 References
- 1. Cystic Fibrosis Australia. Australian Cystic Fibrosis Data Registry Annual Report 2022. North Ryde, NSW: Cystic Fibrosis Australia; 2023.
- 2. Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018;17(2):153–178.
- 3. Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809–1819.
- 4. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial (VX-445-104). Lancet. 2019;394(10212):1940–1948.
- 5. Australian Government Department of Health and Aged Care. Pharmaceutical Benefits Scheme — Elexacaftor with tezacaftor with ivacaftor (Trikafta). Canberra: Commonwealth of Australia; 2022. Available at: pbs.gov.au.
- 6. Döring G, Flume P, Heijerman H, Elborn JS, et al. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros. 2012;11(6):461–479.
- 7. Taccetti G, Bianchini E, et al. Pseudomonas aeruginosa eradication in cystic fibrosis: a consensus statement from the Italian Society of Cystic Fibrosis. J Cyst Fibros. 2020;19(2):195–203.
- 8. Moran A, Brunzell C, Cohen RC, et al. Clinical care guidelines for cystic fibrosis–related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33(12):2697–2708.
- 9. Haworth CS, Bilton D, Elborn JS. Long-term safety and efficacy of inhaled dry powder mannitol in the treatment of cystic fibrosis. J Cyst Fibros. 2014;13(4):418–425.
- 10. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367–416.
- 11. Snijders D, Bengt K, et al. Antibiotic therapies for Staphylococcus aureus lung infections in cystic fibrosis. Cochrane Database Syst Rev. 2017;4:CD006851.
- 12. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023. Canberra: AIHW; 2023.
- 13. NHMRC Centre of Research Excellence in Cystic Fibrosis. Australian CF Clinical Practice Guidelines. Melbourne: Murdoch Children's Research Institute; 2020 (updated 2023).
- 14. Sly PD, Gangell CL, Chen L, et al. Risk of bronchiectasis in cystic fibrosis detected by computed tomography. N Engl J Med. 2013;369(23):2212–2221.
- 15. Dijk FN, McKay K, Barzi F, et al. Improved survival in cystic fibrosis in Australia: the impact of newborn screening and CFTR modulator therapy. J Cyst Fibros. 2022;21(5):767–774.