📋 Key Information Summary
- ADPKD is the most common inherited kidney disease (prevalence ~1:1,000–4,000), caused by pathogenic variants in PKD1 (~78%) or PKD2 (~15%); PKD1 mutations cause more severe disease with ESRF median age 54 v 74 years for PKD2.
- ARPKD is far rarer (~1:20,000–40,000), caused by biallelic PKHD1 mutations, typically presents in utero or in infancy with enlarged kidneys and congenital hepatic fibrosis.
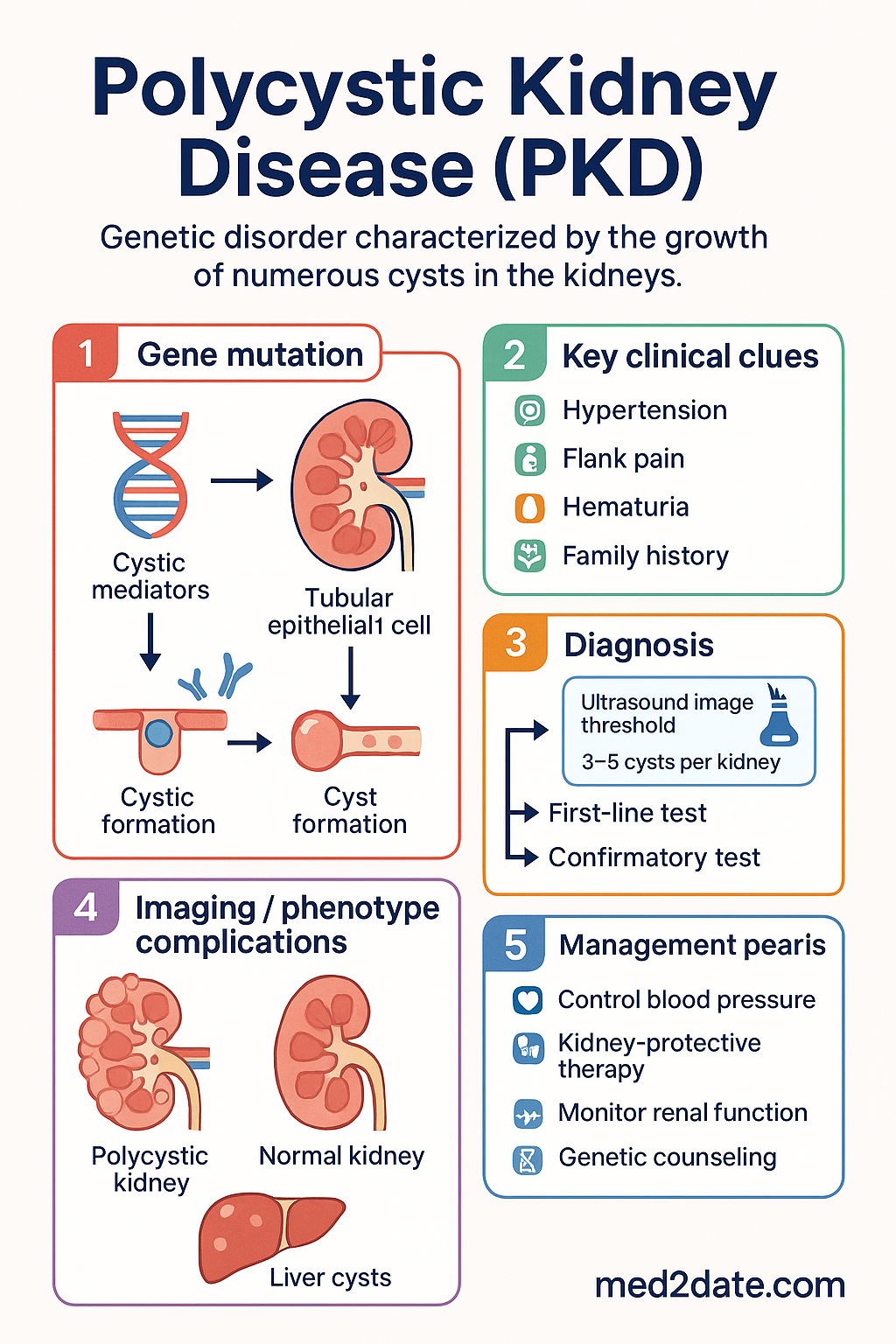
- Diagnosis of ADPKD in at-risk adults (18–39 y): ≥3 unilateral or bilateral renal cysts on ultrasound (Ravine criteria revised by Pei); total kidney volume (TKV) on MRI is the best predictor of progression.
- Extrarenal manifestations include hepatic cysts (83%), intracranial aneurysms (5–10%), mitral valve prolapse (25%), and colonic diverticular disease.
- Target blood pressure <130/80 mmHg for all ADPKD patients; ACE inhibitors or ARBs are first-line antihypertensives.
- Tolvaptan (Jinarc®) slows cyst growth and eGFR decline in rapidly progressive ADPKD; PBS Authority Required listing in Australia with specific criteria.
- Screen for intracranial aneurysm with MRA if family history of SAH or aneurysm, or high-risk occupation; do not routinely screen asymptomatic low-risk patients.
- ~50% of ADPKD patients develop ESRF requiring RRT; causes ~10% of prevalent dialysis and ~6% of kidney transplant recipients in Australia.
- Manage volume status with adequate hydration (≥2.5–3 L/day); avoid excessive sodium intake (<6 g NaCl/day); counsel against nephrotoxic exposures.
- Aboriginal and Torres Strait Islander Australians have later presentation and reduced access to specialist nephrology care and pre-emptive transplantation.
- Genetic counselling is essential for all families; pre-symptomatic screening of at-risk children raises ethical considerations (no disease-modifying therapy for paediatric ADPKD).
- ADPKD accounts for 5–10% of the Australian dialysis population (~1,200 patients); median age at ESRF 54 years for PKD1, 74 years for PKD2.
Introduction & Australian Epidemiology
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease worldwide, caused predominantly by pathogenic variants in PKD1 (chromosome 16p13.3, encoding polycystin-1) and PKD2 (chromosome 4q21, encoding polycystin-2). It is characterised by the progressive bilateral development and growth of renal cysts, leading to a gradual decline in kidney function and eventual end-stage renal failure (ESRF) in approximately 50% of affected individuals.
In Australia, ADPKD affects an estimated 1 in 1,000 to 4,000 persons, with prevalence among the dialysis population of approximately 5–10% according to the Australia and New Zealand Dialysis and Transplant Registry (ANZDATA). At any given time, approximately 1,200 Australians with ADPKD are receiving dialysis or are living with a kidney transplant. The disease imposes significant morbidity, healthcare costs, and psychosocial burden on patients and families across all Australian jurisdictions.
Autosomal recessive polycystic kidney disease (ARPKD) is far rarer, with an estimated incidence of 1:20,000 to 40,000 live births. ARPKD is caused by biallelic mutations in PKHD1 (chromosome 6p12.2, encoding fibrocystin/polyductin) and typically presents antenatally or in early childhood with massively enlarged, echogenic kidneys and congenital hepatic fibrosis.
This guideline addresses both ADPKD and ARPKD, covering genetics, clinical features, extrarenal manifestations, diagnostic investigations, and contemporary management including disease-modifying therapy, blood pressure optimisation, and preparation for renal replacement therapy (RRT).
Genetics: ADPKD vs ARPKD
ADPKD — Autosomal Dominant Inheritance
ADPKD follows autosomal dominant inheritance with near-complete penetrance by age 80. Two major genes account for >90% of cases:
- PKD1 (~78% of families) — encodes polycystin-1, a large transmembrane receptor involved in cell–cell and cell–matrix signalling. PKD1 mutations produce more severe disease: median age at ESRF 54 years.
- PKD2 (~15% of families) — encodes polycystin-2, a non-selective cation channel. PKD2 mutations produce milder disease: median age at ESRF 74 years.
- A minority (~7%) have no identifiable mutation on conventional sequencing; deep intronic variants and mosaic mutations are increasingly recognised.
ARPKD — Autosomal Recessive Inheritance
ARPKD is caused by biallelic pathogenic variants in PKHD1, encoding fibrocystin/polyductin. Key features:
- Both parents are obligate carriers (heterozygous); recurrence risk 25% per pregnancy.
- Carrier frequency ~1:70 in Caucasian populations; homozygous or compound heterozygous state leads to disease.
- Phenotypic spectrum ranges from perinatal lethality (Potter sequence) to presentation in adolescence with portal hypertension due to congenital hepatic fibrosis.
- Approximately 30% of affected neonates die in the perinatal period from pulmonary hypoplasia due to oligohydramnios.
| Feature | ADPKD | ARPKD |
|---|---|---|
| Inheritance | Autosomal dominant | Autosomal recessive |
| Gene | PKD1, PKD2 | PKHD1 |
| Prevalence | 1:1,000–4,000 | 1:20,000–40,000 |
| Typical age at dx | 30–50 years | In utero – infancy |
| Renal cysts | Macroscopic, bilateral, progressive | Microscopic, diffuse collecting-duct dilatation |
| Liver involvement | Simple hepatic cysts (83%) | Congenital hepatic fibrosis (caroli complex) |
| ESRF age (median) | 54 y (PKD1), 74 y (PKD2) | Variable; ~20–30% in childhood |
| Genetic testing | NGS panel / WES | PKHD1 sequencing |
Genetic Testing in Australia
Indications for genetic testing include: (1) atypical imaging (unilateral or asymmetric disease, early-onset, no family history); (2) potential living kidney donor evaluation; (3) reproductive counselling; and (4) paediatric presentations. Testing is available through accredited laboratories in all Australian states (e.g., Victorian Clinical Genetics Services, SA Pathology, PathWest). Costs may be partially offset by Medicare under MBS item 73309 (familial mutation testing) when a known family variant exists. Panel-based next-generation sequencing (NGS) for PKD1/PKD2 costs approximately AUD 500–800 in the private setting.
Clinical Features & Extrarenal Manifestations
Renal Manifestations
- Renal enlargement: Progressive bilateral kidney enlargement detectable by palpation in ~60% of patients by age 40. Total kidney volume (TKV) is the strongest predictor of future GFR decline.
- Flank or abdominal pain: Most common presenting symptom (~60%); due to cyst enlargement, haemorrhage into a cyst, or nephrolithiasis.
- Haematuria: Gross or microscopic; may follow trauma, exercise, or spontaneous cyst rupture.
- Nephrolithiasis: Affects 20–30%; uric acid (57%) and calcium oxalate stones; metabolic workup recommended.
- Urinary tract infections and cyst infections: Increased susceptibility; cyst infection is notoriously difficult to treat (poor drug penetration).
- Hypertension: Present in >60% before any measurable GFR decline; driven by intrarenal activation of the RAAS system.
- ESRF: Develops in ~50%; median age 54 y (PKD1) to 74 y (PKD2).
Extrarenal Manifestations
ARPKD-Specific Features
- Massively enlarged, echogenic kidneys bilaterally on antenatal ultrasound.
- Pulmonary hypoplasia secondary to oligohydramnios — leading cause of perinatal death (~30%).
- Congenital hepatic fibrosis and Caroli disease — portal hypertension, cholangitis, varices.
- Hypertension in >80% of survivors; requires aggressive antihypertensive therapy.
Investigations (Ultrasound, MRI, Genetics)
Imaging Criteria for ADPKD Diagnosis
| Age (years) | Positive diagnosis criteria (ultrasound) | Negative to exclude ADPKD |
|---|---|---|
| 15–39 | ≥3 cysts, unilateral or bilateral | <3 cysts (high negative predictive value) |
| 40–59 | ≥2 cysts in each kidney | <2 cysts in each kidney |
| ≥60 | ≥4 cysts in each kidney | <4 cysts in each kidney (less reliable) |
These revised unified criteria (Pei et al., 2009, 2015) supersede the older Ravine criteria and improve sensitivity for PKD2 carriers. They assume a known family history of ADPKD and the absence of other cystic diseases.
Prognostic Classification (Mayo Classification)
The Mayo Clinic classification stratifies ADPKD patients into five height-adjusted TKV (htTKV) classes based on age and kidney volume, predicting the rate of GFR decline:
Management (Tolvaptan, Blood Pressure, ESRF)
Blood Pressure Management
Hypertension is present in >60% of ADPKD patients before any measurable GFR decline and is associated with faster progression to ESRF, left ventricular hypertrophy, and increased cardiovascular risk.
- Target: <130/80 mmHg for all ADPKD patients (KDIGO 2024, RACGP).
- First-line: ACE inhibitor (e.g., ramipril) or ARB (e.g., telmisartan) — preferred over calcium channel blockers due to superior renoprotective effects and evidence from the HALT-PKD trial.
- Combine with non-dihydropyridine CCB (diltiazem) or diuretic as second-line if target not achieved.
- Avoid excessive diuresis — maintain high fluid intake (≥2.5–3 L/day) to suppress vasopressin-mediated cyst growth.
Tolvaptan — Disease-Modifying Therapy
Tolvaptan (Jinarc®) is a selective vasopressin V2 receptor antagonist that reduces cAMP-driven cyst proliferation and fluid secretion. The TEMPO 3:4 trial demonstrated a 49% reduction in the rate of TKV growth and a 26% reduction in eGFR decline over 3 years. The REPRISE trial confirmed benefit in later-stage ADPKD (eGFR 25–65 mL/min).
Supportive & Preventive Measures
| Measure | Recommendation | Evidence basis |
|---|---|---|
| Fluid intake | ≥2.5–3 L water per day (suppress vasopressin) | Observational; supported by TEMPO rationale |
| Sodium restriction | <6 g NaCl/day (100 mmol Na⁺) | HALT-PKD; reduces HTN and proteinuria |
| Avoid nephrotoxins | Minimise NSAIDs, IV contrast (hydrate if necessary), aminoglycosides | General CKD principles; KDIGO |
| Weight management | BMI 18.5–25 kg/m² | Observational; slower GFR decline |
| Exercise | Regular moderate activity; avoid contact sports (risk of cyst rupture/haemorrhage) | Expert consensus |
| Pregnancy | Safe in most; stop tolvaptan before conception (teratogenicity risk); monitor HTN closely | FDA pregnancy category X (tolvaptan) |
Management of ESRF & Renal Replacement Therapy
Approximately 50% of ADPKD patients will develop ESRF. Preparation for RRT should begin when eGFR falls below 20–25 mL/min.
Management of Cyst Complications
- Cyst infection: Fluoroquinolones (ciprofloxacin 500–750 mg PO BD) are first-line due to superior cyst penetration. Treat for 4–6 weeks. If unresponsive: surgical or percutaneous drainage.
- Cyst haemorrhage: Usually self-limiting; conservative management, adequate hydration, avoid antiplatelet agents acutely.
- Nephrolithiasis: Metabolic workup (serum urate, calcium, citrate; 24-h urine). Treat with increased fluid intake, potassium citrate for hypocitraturia, and standard urological management.
- Chronic pain: Step-wise analgesic approach; avoid long-term NSAIDs. Consider cyst aspiration with sclerotherapy, surgical cyst decortication, or rarely nephrectomy.
Special Populations
Aboriginal and Torres Strait Islander Health
📚 References
- 1. Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015;88(1):17–27.
- 2. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med. 2017;377(20):1930–1942.
- 3. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418. (TEMPO 3:4)
- 4. Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014;371(24):2255–2266. (HALT-PKD Study A)
- 5. Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205–212.
- 6. Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26(1):160–172.
- 7. The National ESKD Advisory Committee and Kidney Health Australia. Chronic Kidney Disease (CKD) Management in Primary Care. 4th edition. Melbourne: Kidney Health Australia; 2020.
- 8. AIHW. Chronic kidney disease in Aboriginal and Torres Strait Islander people. Cat. no. PHE 251. Canberra: Australian Institute of Health and Welfare; 2022.
- 9. ANZDATA Registry. 46th Annual Report 2023 (Data to 2022). Adelaide: Australia and New Zealand Dialysis and Transplant Registry.
- 10. KDIGO 2024 Clinical Practice Guideline for the Evaluation, Management, and Treatment of Chronic Kidney Disease. Kidney Int. 2024;105(4S):S117–S314.
- 11. Chebib FT, Torres VE. Autosomal dominant polycystic kidney disease: core curriculum 2016. Am J Kidney Dis. 2016;67(5):792–810.
- 12. Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4(1):50.
- 13. Horie S, Mochizuki T, Kaname S. Autosomal dominant polycystic kidney disease: pathogenesis, biomarkers, and treatment. Clin Exp Nephrol. 2022;26(7):623–634.
- 14. Services Australia. Pharmaceutical Benefits Schedule — Tolvaptan. PBS Item 12324Y. Canberra; 2024. Available from: pbs.gov.au.