📋 Key Information Summary
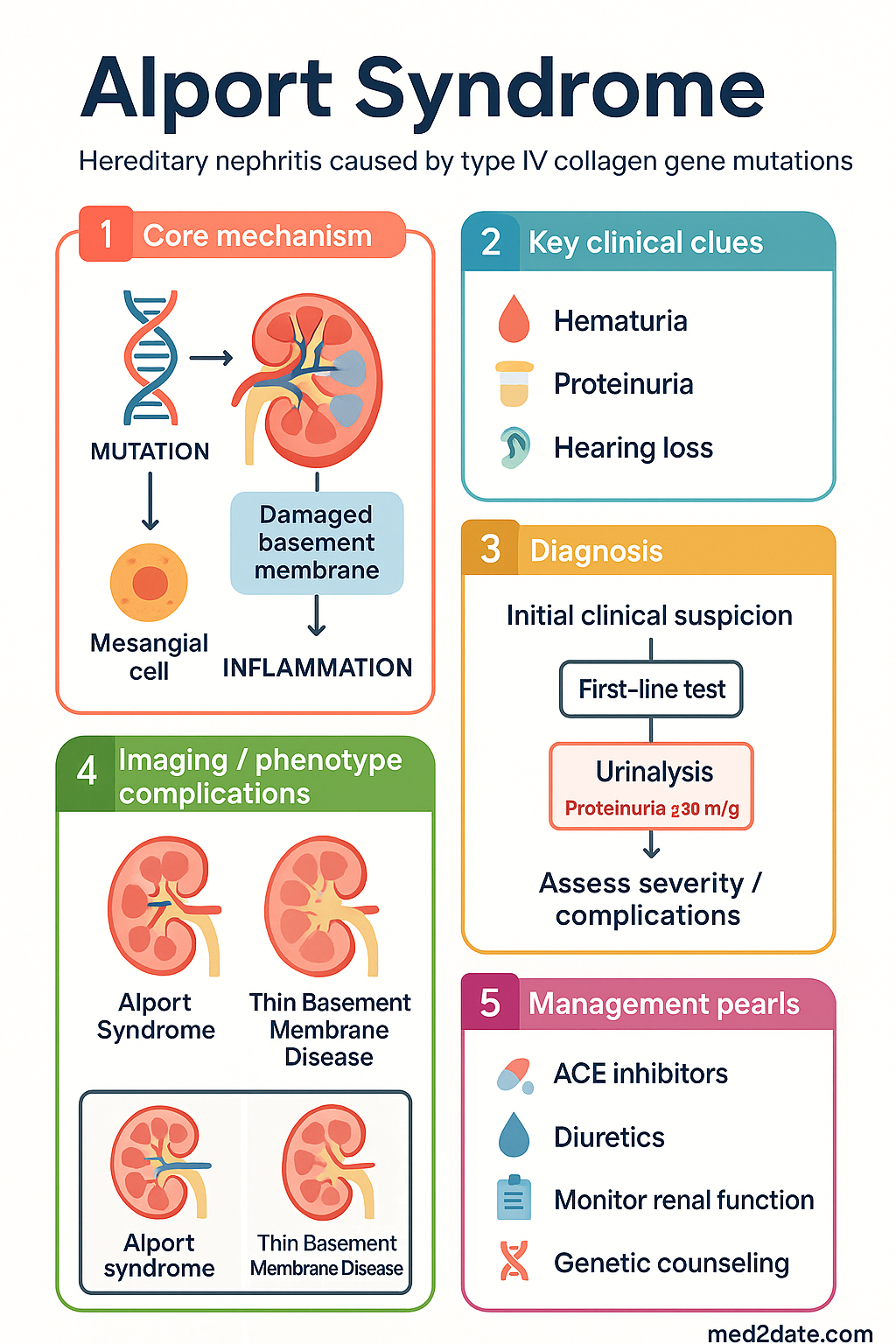
- Definition: Alport syndrome is a hereditary nephritis caused by pathogenic variants in type IV collagen genes (COL4A3, COL4A4, COL4A5), leading to progressive kidney disease, sensorineural hearing loss, and ocular abnormalities.
- Inheritance: X-linked (80%, COL4A5), autosomal recessive (15%, COL4A3/COL4A4), and autosomal dominant (5%) forms.
- Clinical Triad: Persistent microscopic haematuria (always present), progressive sensorineural hearing loss (high-frequency), and anterior lenticonus (pathognomonic).
- Renal Outcome: Progressive chronic kidney disease (CKD) leading to end-stage kidney disease (ESKD) in most males with X-linked disease by age 30.
- Diagnostic Investigations: Skin biopsy for α5(IV) collagen (X-linked), renal biopsy showing thinning and lamellation of glomerular basement membrane (GBM), and genetic testing (definitive).
- Key Management Principle: Early and sustained renin-angiotensin-aldosterone system (RAAS) blockade with ACE inhibitors (ACEi) or ARBs to delay progression to ESKD.
- Transplant Outcomes: Renal transplantation is the treatment for ESKD; outcomes are excellent, though ~3-5% of males may develop anti-GBM disease post-transplant.
- Monitoring: Annual audiology and ophthalmology reviews from age 6-10 years, alongside regular renal function and proteinuria monitoring.
- Genetic Counselling: Essential for all families due to hereditary nature and implications for family planning.
- Australian Considerations: Genetic testing is available via specialist genetic services and may be Medicare-rebatable under specific MBS items; multidisciplinary care is essential.
Introduction & Australian Epidemiology
Alport syndrome is a progressive hereditary nephropathy characterised by a classic triad of haematuria, sensorineural deafness, and ocular abnormalities. It accounts for approximately 1-2% of ESKD in Australia and up to 5% of children undergoing renal transplantation. The condition is caused by mutations in the genes encoding type IV collagen, a critical structural component of basement membranes. Early diagnosis and intervention with RAAS inhibitors are paramount to preserving renal function. The spectrum of disease severity and extra-renal manifestations varies significantly with the specific genetic defect and inheritance pattern.
Genetics & Pathophysiology (Type IV Collagen)
Alport syndrome results from pathogenic variants in the genes COL4A3, COL4A4, and COL4A5. These genes encode the α3, α4, and α5 chains of type IV collagen, which form the essential structural network in the glomerular basement membrane (GBM), the basement membrane of the cochlea (stria vascularis), and the lens capsule.
| Inheritance Pattern | Gene(s) Affected | Proportion | Typical Severity |
|---|---|---|---|
| X-linked (XLAS) | COL4A5 | ~80% | Males: severe; Females: variable (carrier status) |
| Autosomal Recessive (ARAS) | COL4A3 or COL4A4 | ~15% | Severe, similar to XLAS in males |
| Autosomal Dominant (ADAS) | COL4A3 or COL4A4 | ~5% | Often milder, slower progression |
Pathophysiological Sequence: Mutations lead to abnormal α345(IV) collagen trimers. This results in defective GBM assembly, instability, and progressive splitting/lamellation visible on electron microscopy. The defective cochlear and lens capsule basement membranes underpin the extra-renal manifestations.
Clinical Features
Presentation varies by inheritance pattern, sex, and age. The classic triad is pathognomonic but not universally present at diagnosis.
- Persistent Microscopic Haematuria: Universal and often the first sign, present from birth in males with XLAS and ARAS.
- Proteinuria: Develops in childhood and is progressive; a key marker of disease progression.
- Hypertension: Develops with advancing CKD.
- Progressive CKD: Leads to ESKD, typically by age 16-35 in males with XLAS, and later in ADAS.
- Sensorineural Hearing Loss: Bilateral, high-frequency deficit. Typically develops in late childhood (age 10+) and is progressive. Not congenital.
- Ocular Abnormalities:
- Anterior Lenticonus: Pathognomonic cone-shaped protrusion of the lens central pole.
- Dot-and-fleck retinopathy, macular hypoplasia, posterior polymorphous corneal dystrophy.
- Leiomyomatosis: Rare, associated with contiguous deletions of COL4A5/COL4A6 (XLAS).
Investigations
A combination of histopathology and genetic testing establishes the diagnosis. Genetic testing is becoming the gold standard.
Management (ACEi, Renal Transplant)
There is no cure. Management focuses on slowing progression to ESKD, managing complications, and preparing for renal replacement therapy.
Pharmacological Therapy (RAAS Blockade)
Supportive & Preventive Care
- Blood Pressure Control: Target <130/80 mmHg (or age-appropriate in paediatrics).
- Dietary Advice: Sodium restriction. Moderate protein intake (no restriction unless advanced CKD).
- Nephrotoxin Avoidance: NSAIDs, aminoglycosides. Judicious use of contrast media.
- Hearing Aids: For progressive hearing loss. Referral to audiologist.
- Vaccination: Ensure pneumococcal and hepatitis B (pre-transplant) vaccines are up to date.
Renal Replacement Therapy & Transplantation
- Living Related Donation: Requires careful genetic screening of potential related donors to exclude affected individuals (carriers of XLAS or ARAS).
- Post-Transplant Monitoring: Monitor for anti-GBM disease (~3-5% of transplanted males with XLAS), which can cause rapid graft loss. Presents with haematuria and rising creatinine.
- Dialysis: Haemodialysis or peritoneal dialysis as bridging therapy.
Specific prevalence data for Alport syndrome in Aboriginal and Torres Strait Islander communities is limited. However, general principles of equitable care, genetic counselling access, and managing CKD complications in the context of remote or rural settings are paramount.
📚 References
- 1. Kashtan CE, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345. J Am Soc Nephrol. 2018;29(7):1937-1947.
- 2. Savige J, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013;24(3):364-375.
- 3. Kidney Health Australia. Alport Syndrome - Fact Sheet for Health Professionals. 2022.
- 4. Royal Australasian College of Physicians (RACP). Caring for Australasians with Alport syndrome: A consensus statement. 2020.
- 5. Gross O, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81(6):494-501.
- 6. Australian Institute of Health and Welfare (AIHW). Kidney disease in Aboriginal and Torres Strait Islander people. Cat. no. PHE 219. Canberra: AIHW; 2018.
- 7. Mallett A, et al. Genomic testing in the diagnosis of nephropathy: a national perspective. Nephrology. 2021;26(5):414-422.
- 8. Transplant Society of Australia and New Zealand (TSANZ). Clinical Guidelines for Kidney Transplantation. 2023.
- 9. Jais JP, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol. 2003;14(10):2603-2610.
- 10. Rheault MN. Women and Alport syndrome. Pediatr Nephrol. 2012;27(1):41-46.
- 11. Genetic Health Service Australasia (GHSA). Guidelines for Genetic Testing of Heritable Renal Disorders. 2021.