📋 Key Information Summary
- Bartter syndrome, Gitelman syndrome, and Liddle syndrome are hereditary tubulopathies presenting with hypokalaemia and metabolic alkalosis, but differ fundamentally in mechanism, salt-wasting vs. salt-retention, and blood pressure.
- Bartter syndrome: Loop of Henle defect causing salt wasting, hypotension/hypotension, hypokalaemic alkalosis, hypercalciuria, and elevated renin–aldosterone with normal/low BP.
- Gitelman syndrome: Distal convoluted tubule (DCT) SLC12A3 defect — milder presentation, hypomagnesaemia, hypocalciuria, often diagnosed in adolescence/adulthood.
- Liddle syndrome: Gain-of-function mutations in SCNN1B/SCNN1G causing constitutive ENaC activation — severe hypertension with suppressed renin and aldosterone; hypokalaemia from ENaC-mediated potassium wasting.
- Antenatal Bartter syndrome (types I–III) presents with polyhydramnios, prematurity, and life-threatening neonatal salt wasting; classic Bartter (type III) is milder.
- Gitelman syndrome is the most common inherited salt-losing tubulopathy (prevalence ~1:40,000) and is frequently diagnosed incidentally.
- Liddle syndrome is a rare Mendelian form of early-onset hypertension — amiloride/triamterene are specific therapies; spironolactone is ineffective.
- Key differentiating investigation: 24-hour urine calcium (high in Bartter, low in Gitelman) and plasma renin–aldosterone pattern (high in Bartter/Gitelman, suppressed in Liddle).
- Bartter and Gitelman require potassium and magnesium replacement; Bartter additionally needs prostaglandin synthase inhibitors (indomethacin, celecoxib).
- Liddle syndrome requires amiloride or triamterene (direct ENaC blockade); spironolactone and eplerenone are NOT effective.
- Genetic confirmation via next-generation sequencing panels (SLC12A1, KCNJ1, CLCNKB, BSND, SLC12A3, SCNN1A/B/G) is recommended — available at major Australian genetics laboratories.
- All three conditions predispose to growth impairment in children; Bartter/Gitelman may cause nephrocalcinosis and chronic kidney disease.
- In Aboriginal and Torres Strait Islander communities, access to specialist genetics services and ongoing electrolyte monitoring is limited in remote areas; telehealth nephrology outreach is essential.
Introduction & Australian Epidemiology
Bartter syndrome, Gitelman syndrome, and Liddle syndrome constitute a clinically important group of inherited renal tubulopathies characterised by disordered sodium, potassium, and acid–base handling. Despite sharing the unifying presentation of hypokalaemia with metabolic alkalosis, these conditions arise from distinct genetic defects along the nephron and diverge fundamentally in their haemodynamic profiles. Bartter and Gitelman syndromes are salt-wasting disorders associated with normal or low blood pressure, secondary hyperreninaemic hyperaldosteronism, and chronic electrolyte depletion. In contrast, Liddle syndrome is a salt-retaining disorder manifesting as severe early-onset hypertension with suppressed renin and aldosterone.
In Australia, precise incidence data for these rare conditions are limited. Gitelman syndrome is the most prevalent, estimated at approximately 1 in 40,000 individuals globally, with Bartter syndrome (all subtypes combined) estimated at 1 in 1,000,000. Liddle syndrome is extremely rare, with fewer than 100 families reported worldwide, though underdiagnosis is likely given the phenotypic overlap with essential hypertension. Genetic testing in Australia has improved diagnostic rates, with next-generation sequencing (NGS) panels available through state genetics services and commercial laboratories.
These disorders are encountered across all age groups. Antenatal and neonatal forms of Bartter syndrome (types I, II, and IV) present with polyhydramnios, prematurity, and life-threatening salt wasting in the neonatal period. Classic Bartter syndrome (type III) and Gitelman syndrome typically present in childhood or adolescence, while Liddle syndrome may present at any age with treatment-resistant hypertension. Recognising these conditions is critical, as specific therapies differ: prostaglandin inhibitors for Bartter, electrolyte repletion for Gitelman, and amiloride for Liddle — spironolactone is ineffective in Liddle syndrome and may delay correct diagnosis.
Bartter Syndrome: Genetics & Clinical Features
Bartter syndrome encompasses a group of autosomal recessive disorders resulting from defective sodium chloride reabsorption in the thick ascending limb (TAL) of the loop of Henle. Five molecular subtypes are recognised, each reflecting the specific transporter or channel affected.
| Type | Gene | Protein / Function | Typical Onset | Key Features |
|---|---|---|---|---|
| I | SLC12A1 | NKCC2 (Na⁺–K⁺–2Cl⁻ cotransporter) | Antenatal / neonatal | Polyhydramnios, prematurity, hypercalciuria, nephrocalcinosis |
| II | KCNJ1 | ROMK (renal outer medullary K⁺ channel) | Antenatal / neonatal | Similar to Type I; transient hyperkalaemia may occur in first weeks of life |
| III | CLCNKB | ClC-Kb (basolateral chloride channel) | Childhood / adolescence | 'Classic' Bartter; milder; variable hypercalciuria; nephrocalcinosis less common |
| IVa | BSND | Barttin (accessory subunit of ClC-Kb/Ka) | Antenatal / neonatal | Severe antenatal form; sensorineural deafness |
| IVb | CLCNKA + CLCNKB (digenic) | ClC-Ka + ClC-Kb | Antenatal / neonatal | Similar to IVa; sensorineural deafness |
Clinical Features
Antenatal/neonatal Bartter (types I, II, IV): Polyhydramnios due to fetal polyuria, often leading to premature delivery. Neonatal presentation includes severe polyuria, salt wasting with dehydration, failure to thrive, and potentially life-threatening hypokalaemia and metabolic alkalosis. Hypercalciuria is prominent, and bilateral nephrocalcinosis develops in the first year of life in most cases. Type IV is additionally characterised by sensorineural deafness.
Classic Bartter (type III): Presents in mid-childhood to adolescence with polyuria, polydipsia, salt craving, muscle weakness, cramps, growth retardation, and episodes of dehydration. Blood pressure is normal or low. Hypercalciuria is variable, and nephrocalcinosis occurs in approximately 50% of patients.
Biochemical Hallmarks
- Hypokalaemia (typically 2.0–3.0 mmol/L)
- Metabolic alkalosis (elevated bicarbonate)
- Elevated plasma renin activity (PRA) and aldosterone
- Hypercalciuria (24-hour urine calcium/creatinine ratio elevated)
- Normal or low blood pressure
- Elevated urinary prostaglandin E₂ (PGE₂) — particularly in neonatal forms
- Hypomagnesaemia may occur but is less prominent than in Gitelman
Gitelman Syndrome: Genetics & Clinical Features
Gitelman syndrome is an autosomal recessive tubulopathy caused by inactivating mutations in the SLC12A3 gene, encoding the thiazide-sensitive sodium-chloride cotransporter (NCC) in the distal convoluted tubule (DCT). It is the most common inherited salt-losing tubulopathy, with an estimated prevalence of 1 in 40,000 and a carrier frequency of approximately 1% in European populations. Over 500 pathogenic variants have been described.
Genetics
SLC12A3 (chromosome 16q13) is the sole gene causative for Gitelman syndrome in the vast majority of cases. Rarely, mutations in CLCNKB (also responsible for Bartter type III) can produce a Gitelman-like phenotype. Inheritance is autosomal recessive; compound heterozygosity is common. Genetic testing via NGS tubulopathy panels is available through Australian clinical genetics services (e.g., Victorian Clinical Genetics Services, NSW Health Pathology Genetics).
Clinical Features
Gitelman syndrome generally presents later than Bartter syndrome, typically in late childhood, adolescence, or early adulthood. Many patients are diagnosed incidentally following routine blood tests. Presentation is characteristically milder than Bartter syndrome.
- Asymptomatic hypokalaemia: The most common presentation; discovered incidentally.
- Symptomatic hypokalaemia: Muscle weakness, cramps, fatigue, paraesthesias, tetany, carpopedal spasm, and rarely rhabdomyolysis or paralysis.
- Hypomagnesaemia: Present in the majority of patients and often severe (0.4–0.6 mmol/L); this is a key distinguishing feature from Bartter syndrome.
- Salt craving: Preference for salty foods; polydipsia and polyuria are less prominent than in Bartter syndrome.
- Normal or low blood pressure: Despite elevated renin and aldosterone.
- Growth and puberty: Usually normal; growth retardation is uncommon.
- Chondrocalcinosis: Reported in adult patients with longstanding hypomagnesaemia.
- Cardiac: Prolonged QTc interval (due to hypokalaemia and hypomagnesaemia); rare but potentially fatal arrhythmias.
Biochemical Hallmarks
- Hypokalaemia (typically 2.5–3.5 mmol/L; milder than Bartter)
- Metabolic alkalosis
- Hypocalciuria — key differentiator from Bartter syndrome (24-hour urine calcium <0.5 mmol/day or urine Ca/Cr ratio low)
- Hypomagnesaemia — often more severe than in Bartter
- Elevated plasma renin and aldosterone (moderate elevation)
- Normal or low blood pressure
Liddle Syndrome: Pathophysiology & Management
Pathophysiology
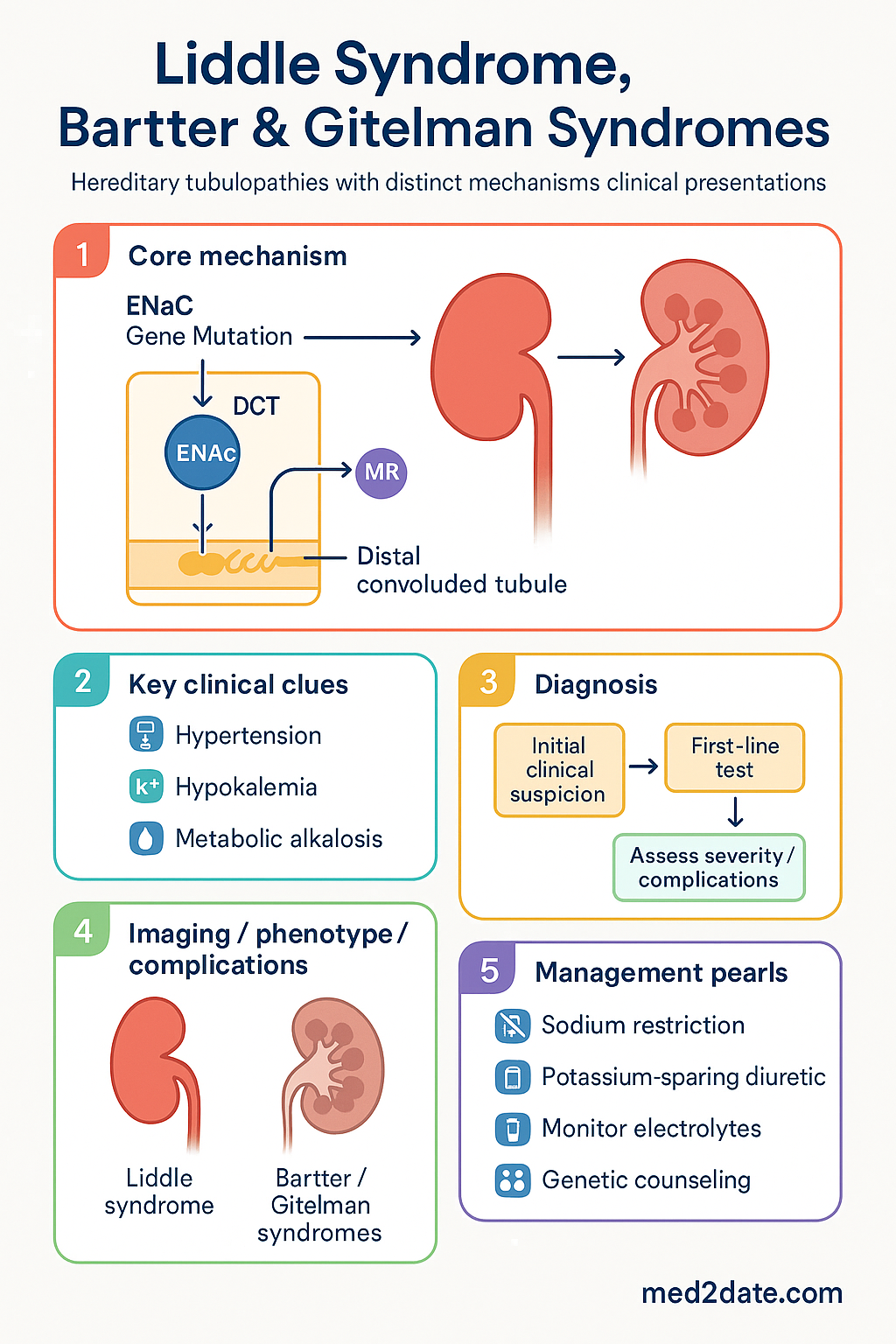
Liddle syndrome is an autosomal dominant disorder caused by gain-of-function mutations in genes encoding subunits of the epithelial sodium channel (ENaC) in the distal nephron. The affected genes are SCNN1B (β-subunit) and SCNN1G (γ-subunit), with rare mutations in SCNN1A (α-subunit). These mutations truncate or alter the proline-rich PY motif in the C-terminus of the ENaC subunits, preventing binding by the ubiquitin ligase Nedd4-2, which normally targets ENaC for endocytosis and degradation.
As a result, ENaC channels accumulate on the apical membrane of the collecting duct principal cell in a constitutively open state. This leads to:
- Excessive sodium reabsorption: Volume expansion and hypertension.
- Intraluminal electronegativity: Enhanced K⁺ and H⁺ secretion into the tubular lumen, causing hypokalaemia and metabolic alkalosis.
- Suppressed renin–aldosterone: Plasma renin activity and aldosterone levels are markedly suppressed (or undetectable) due to volume expansion — a critical distinguishing feature from Bartter and Gitelman syndromes.
- The hypertension is salt-sensitive and unresponsive to mineralocorticoid receptor antagonists (spironolactone, eplerenone) because the defect is downstream of aldosterone.
Clinical Presentation
Liddle syndrome presents with early-onset, severe, treatment-resistant hypertension. Key clinical features include:
- Hypertension presenting in childhood, adolescence, or young adulthood (often before age 30).
- Family history of early-onset hypertension and/or stroke.
- Hypokalaemia (may be mild or absent in some patients).
- Metabolic alkalosis.
- Low or suppressed plasma renin and aldosterone.
- Absence of oedema (despite sodium retention, compensatory mechanisms limit overt oedema).
- Left ventricular hypertrophy and end-organ damage may occur early if untreated.
- No response to spironolactone.
Management
Adjunctive Measures
- Sodium restriction: Dietary salt restriction (<100 mmol Na⁺/day) augments the effect of amiloride.
- Additional antihypertensives: If BP remains uncontrolled on amiloride alone, add a thiazide diuretic (synergistic with ENaC blockade), ACE inhibitor, ARB, or calcium channel blocker.
- Genetic counselling: Autosomal dominant inheritance; first-degree relatives should be screened. Genetic testing is available through Australian clinical genetics laboratories.
- Cardiovascular risk management: Standard assessment and management of end-organ damage (echocardiography, renal function, fundoscopy).
Differential Diagnosis of Hypokalaemic Alkalosis
Hypokalaemic metabolic alkalosis is a common clinical scenario with a broad differential. The inherited tubulopathies must be distinguished from acquired causes, particularly diuretic abuse and surreptitious vomiting. A systematic approach integrating blood pressure, renin–aldosterone status, and urinary calcium and magnesium excretion is essential.
| Condition | BP | Renin | Aldosterone | Urine Ca²⁺ | Mg²⁺ | Key Distinguisher |
|---|---|---|---|---|---|---|
| Bartter syndrome | ↓/Normal | ↑↑ | ↑↑ | ↑↑ (hypercalciuria) | Normal/↓ | Genetic; loop diuretic–like physiology |
| Gitelman syndrome | ↓/Normal | ↑ | ↑ | ↓↓ (hypocalciuria) | ↓↓ | Thiazide-like; hypomagnesaemia prominent |
| Liddle syndrome | ↑↑↑ | ↓↓ | ↓↓ | Normal | Normal | ENaC gain-of-function; amiloride-responsive |
| Primary hyperaldosteronism | ↑↑ | ↓↓ | ↑↑ | Normal | Normal/↓ | Aldosterone:renin ratio elevated; adrenal adenoma/bilateral hyperplasia |
| Diuretic abuse (loop) | ↓/Normal | ↑↑ | ↑↑ | ↑ (during effect) | ↓ | Mimics Bartter; urine diuretic screen |
| Diuretic abuse (thiazide) | ↓/Normal | ↑ | ↑ | ↓ | ↓ | Mimics Gitelman; urine diuretic screen |
| Surreptitious vomiting | ↓/Normal | ↑ | ↑ (if Cl⁻ low) | Variable | Normal | Low urine Cl⁻ (<20 mmol/L); high urine pH |
| Apparent mineralocorticoid excess (AME) | ↑↑↑ | ↓↓ | ↓↓ | Normal | Normal | 11β-HSD2 deficiency; cortisol activates MR; liquorice ingestion phenocopy |
| Liquorice excess | ↑↑ | ↓ | ↓ | Normal | Normal | Dietary history; glycyrrhizic acid inhibits 11β-HSD2; reversible |
Diagnostic Algorithm
Investigations
Management & Therapy
Bartter Syndrome
Management aims to correct electrolyte abnormalities, reduce urinary prostaglandin E₂, and support growth in children. Therapy is supportive and lifelong.
Gitelman Syndrome
Management focuses on potassium and magnesium repletion. Most patients require lifelong supplementation.
Summary: Bartter vs. Gitelman vs. Liddle Therapy
Monitoring
Lifelong monitoring is required for all three conditions. Frequency depends on disease severity and stability.
| Parameter | Frequency | Purpose |
|---|---|---|
| Serum K⁺, Na⁺, Cl⁻, HCO₃⁻ | Every 3–6 months (stable); weekly–monthly during titration or in neonates | Detect hypokalaemia, alkalosis; guide supplementation |
| Serum Mg²⁺ | Every 3–6 months (Gitelman); every 6–12 months (Bartter) | Guide Mg²⁺ supplementation; QTc prolongation risk |
| Serum creatinine / eGFR | Every 6–12 months | Monitor CKD progression; NSAID nephrotoxicity in Bartter |
| Blood pressure | Every visit | Liddle: target <130/80 mmHg; Bartter/Gitelman: detect hypotension |
| Growth parameters (children) | Every 3–6 months | Detect growth faltering; adjust therapy |
| ECG (QTc) | Annually; more frequently if symptomatic | Arrhythmia risk from hypokalaemia/hypomagnesaemia |
| Renal ultrasound | At diagnosis then every 1–2 years (Bartter) | Screen for nephrocalcinosis and nephrolithiasis |
| 24-hour urine Ca²⁺ | Annually (Bartter) | Monitor nephrocalcinosis risk |
| Echocardiography (Liddle) | At diagnosis then as clinically indicated | LVH assessment; end-organ damage |
Special Populations
Aboriginal and Torres Strait Islander Health
📚 References
- 1. Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatric Nephrology. 2011;26(10):1789–1802.
- 2. Blanchard A, Bockenhauer D, Bover J, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney International. 2017;91(1):24–33.
- 3. Konrad M, Nijenhuis T, Ariceta G, et al. Diagnosis and management of Bartter syndrome: executive summary of the consensus and guidelines. Nephrology Dialysis Transplantation. 2021;36(3):435–445.
- 4. Salih M, Bovée DM, van der Lubbe N, et al. Liddle syndrome: from genetics to clinical practice. Nephron. 2022;146(4):343–353.
- 5. Ji W, Foo JN, O'Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nature Genetics. 2008;40(5):592–599.
- 6. Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman syndrome. Journal of the American Society of Nephrology. 2011;22(4):693–703.
- 7. Riancho-Zarrabeitia L, Martín-Carmona J, Baranda JC. Bartter and Gitelman syndromes: a practical approach to diagnosis and treatment. Medicina Clínica. 2023;160(8):360–367.
- 8. Australian Institute of Health and Welfare (AIHW). Chronic kidney disease prevalence among Aboriginal and Torres Strait Islander people. AIHW Bulletin 149. Canberra: AIHW; 2023.
- 9. Kidney Health Australia. Chronic kidney disease management in primary care. 4th ed. Melbourne: Kidney Health Australia; 2020.
- 10. Simmonds EJ, Halket S, Devriendt K, et al. Neonatal Bartter syndrome: a case series and review of the literature. Archives of Disease in Childhood — Fetal and Neonatal Edition. 2023;108(2):F161–F166.
- 11. Pharmaceutical Benefits Scheme (PBS). Australian Government Department of Health. Available at: https://www.pbs.gov.au. Accessed 2024.
- 12. Medical Services Advisory Committee (MSAC). Genomic testing under MBS. Australian Government Department of Health; 2023.