📋 Key Information Summary
- Fabry disease is an X-linked lysosomal storage disorder caused by deficiency of alpha-galactosidase A (α-Gal A), leading to progressive accumulation of globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3) in multiple organ systems.
- Classic Fabry disease presents in childhood–adolescence with acroparaesthesia, angiokeratoma, hypohidrosis, and corneal verticillata; end-organ damage (renal, cardiac, cerebrovascular) typically manifests from the third decade.
- Late-onset variants (cardiac or renal predominant) may present in the fifth–seventh decades with isolated left ventricular hypertrophy or proteinuric CKD, often mimicking hypertensive or diabetic nephropathy.
- Renal involvement begins as a Gb3-laden podocytopathy with progressive proteinuria, followed by tubulointerstitial fibrosis and declining eGFR; without treatment, end-stage kidney disease occurs in classically affected males by age 40–50.
- Cardiac manifestations include progressive left ventricular hypertrophy, arrhythmias (atrial fibrillation, ventricular tachycardia), and accelerated coronary artery disease; cardiac death is the leading cause of mortality in females.
- Cerebrovascular events (ischaemic stroke, TIA, vertebrobasilar dolichoectasia) occur at younger ages than the general population; white matter lesions on MRI are common even in adolescents.
- Diagnosis requires α-Gal A enzyme assay (in males) and/or GLA gene sequencing (essential in females due to random X-inactivation); plasma lyso-Gb3 is a sensitive biomarker for disease activity.
- Enzyme replacement therapy (ERT) with agalsidase alfa (Replagal®) or agalsidase beta (Fabrazyme®) is PBS-listed in Australia under Section 100 (Special Authority) for patients meeting specific criteria.
- Migalastat (Galafold®), an oral pharmacological chaperone, is PBS-listed for amenable GLA mutations and offers an alternative to fortnightly intravenous infusions.
- Adjunctive renoprotective therapy with ACE inhibitors or ARBs plus lifestyle modification is critical to slow CKD progression; target proteinuria <500 mg/day.
- Multidisciplinary care in a specialist Fabry centre is recommended; in Australia, dedicated services operate through major tertiary hospitals in Sydney, Melbourne, Brisbane, Adelaide, and Perth.
- Aboriginal and Torres Strait Islander Australians with Fabry disease face significant barriers to diagnosis and treatment access, including remoteness from specialist centres and reduced genetic testing availability.
Introduction & Australian Epidemiology
Fabry disease (OMIM #301500) is an X-linked lysosomal storage disorder caused by pathogenic variants in the GLA gene (Xq22.1), resulting in deficient or absent activity of the enzyme alpha-galactosidase A (α-Gal A, EC 3.2.1.22). The enzymatic defect leads to progressive intracellular accumulation of globotriaosylceramide (Gb3) and its deacylated derivative globotriaosylsphingosine (lyso-Gb3) in endothelial cells, cardiomyocytes, renal podocytes, dorsal root ganglia, and corneal epithelium.
The classic phenotype presents in childhood with debilitating neuropathic pain (acroparaesthesia), angiokeratomas, hypohidrosis, and characteristic corneal opacities (corneal verticillata or vortex keratopathy). Without intervention, progressive Gb3 deposition drives irreversible end-organ damage — particularly in the kidneys, heart, and cerebrovasculature — culminating in end-stage kidney disease (ESKD), heart failure, and premature stroke.
In Australia, Fabry disease is estimated to affect approximately 1 in 40,000–117,000 live births for the classic form, though the true prevalence may be higher due to under-diagnosis of late-onset variants. The Australian Fabry Disease Registry and the Lysosomal Diseases Network have documented several hundred patients nationally, with the largest cohorts in New South Wales and Victoria. Newborn screening pilot programmes have identified a higher-than-expected incidence of GLA variants of uncertain significance, highlighting the diagnostic challenge of late-onset disease.
This guideline provides an Australian-focused clinical framework for the renal physician managing Fabry disease, encompassing genetic and pathological mechanisms, renal and extrarenal manifestations, diagnostic investigation, and evidence-based treatment including enzyme replacement therapy and pharmacological chaperone therapy.
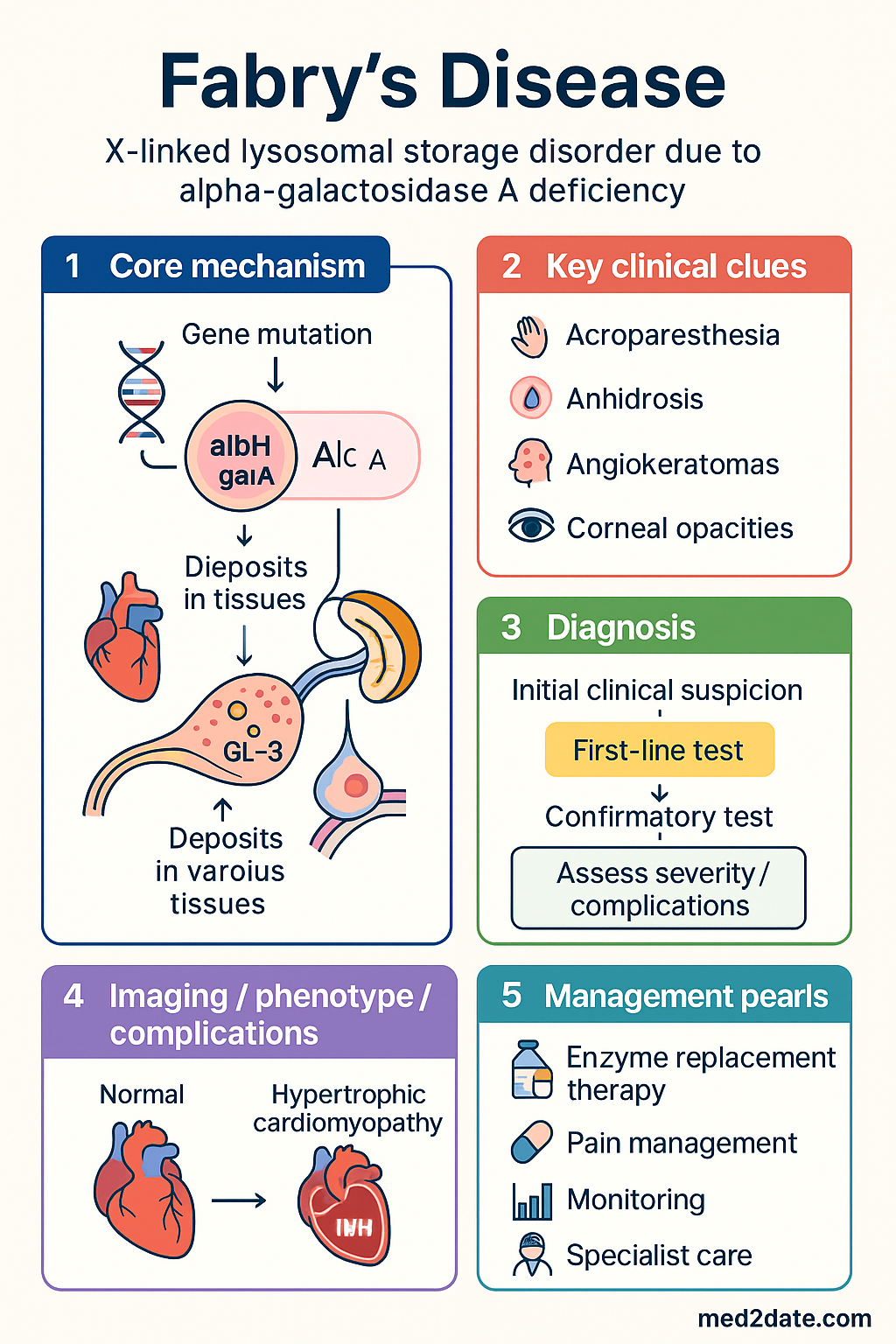
Genetics & Pathophysiology (Gb3 Accumulation)
Genetic Basis
The GLA gene spans approximately 12 kb on chromosome Xq22.1 and encodes the 429-amino-acid enzyme α-Gal A. Over 1,000 pathogenic variants have been catalogued, including missense mutations (~60%), nonsense mutations, splice-site alterations, small insertions/deletions, and large genomic rearrangements. The mutation type largely determines residual enzyme activity and disease phenotype.
| Feature | Classic Fabry | Late-Onset Variant |
|---|---|---|
| Residual α-Gal A activity | <1–3% of normal | 2–30% of normal |
| Onset of symptoms | Childhood (4–8 years) | 30–70 years |
| Classic triad | Present (pain, angiokeratoma, hypohidrosis) | Absent or attenuated |
| Organ involvement | Multisystem (renal, cardiac, CNS) | Predominantly cardiac OR renal |
| Inheritance pattern | X-linked | X-linked (variable expressivity in females) |
| Common variants | Nonsense, frameshift, canonical splice | p.N215S, p.R301Q, IVS4+919G→A |
X-Linkage & Female Carriers
As an X-linked condition, classically affected hemizygous males manifest the full disease spectrum. Heterozygous females, however, are not simply "carriers" — random X-chromosome inactivation (lyonisation) results in a mosaic pattern of α-Gal A expression. Approximately 60–70% of heterozygous females develop clinically significant organ involvement, though onset is typically later and progression slower than in males. Severe Fabry phenotypes in females, including ESKD and stroke, are well documented and mandate full clinical evaluation and treatment consideration.
Pathophysiology of Gb3 Accumulation
α-Gal A cleaves the terminal α-D-galactose residue from globotriaosylceramide (Gb3), a glycosphingolipid derived from the degradation of cell membranes. Deficient enzyme activity causes Gb3 to accumulate within lysosomes of:
- Renal cells: Podocytes, mesangial cells, distal tubular epithelium, and peritubular capillary endothelium — driving the hallmark podocytopathy and progressive glomerulosclerosis.
- Cardiomyocytes: Leading to myocyte hypertrophy, disarray, and progressive fibrosis with concentric left ventricular hypertrophy.
- Endothelial cells: Systemic vasculopathy contributing to cerebral small vessel disease, ischaemic events, and peripheral vascular dysfunction.
- Dorsal root ganglia & autonomic neurons: Neuropathic pain, impaired thermal regulation, and gastrointestinal dysmotility.
Role of Lyso-Gb3
Globotriaosylsphingosine (lyso-Gb3), the deacylated derivative of Gb3, is water-soluble and detectable in plasma. Lyso-Gb3 is more strongly correlated with disease severity than Gb3 itself and promotes fibrogenesis, smooth muscle proliferation, and podocyte injury. Plasma lyso-Gb3 is a valuable biomarker for disease monitoring and treatment response; levels are markedly elevated in classically affected males (>15 nmol/L) and variably elevated in heterozygous females and late-onset variants.
Renal Features (Podocytopathy, CKD)
The kidney is a primary target organ in Fabry disease. Gb3 accumulation within renal cells drives a progressive nephropathy that follows a predictable clinical trajectory, particularly in classically affected males.
Pathological Sequence
Renal pathology in Fabry disease progresses through several stages:
- Subclinical Gb3 deposition (0–10 years): Lysosomal inclusions ("zebra bodies" on electron microscopy) accumulate in podocytes, mesangial cells, and distal tubular epithelium before any clinical or laboratory abnormality is apparent. Urinary Gb3 excretion may be elevated.
- Podocytopathy with microalbuminuria (10–20 years): Podocyte injury triggers glomerular albumin leak. The histological pattern is a distinctive "Fabry nephropathy" with segmental glomerulosclerosis, vacuolated podocytes, and arteriolar hyalinosis — often misdiagnosed as focal segmental glomerulosclerosis (FSGS) or hypertensive nephrosclerosis on light microscopy alone.
- Overt proteinuria and declining eGFR (20–40 years): Progressive glomerulosclerosis and tubulointerstitial fibrosis lead to frank proteinuria (>500 mg/day), hypertension, and a steady decline in eGFR. The rate of eGFR decline in untreated classically affected males averages 5–7 mL/min/1.73 m² per year.
- End-stage kidney disease (40–50 years): Without treatment, ESKD develops in approximately 50% of classically affected males by their mid-forties and in a proportion of heterozygous females, typically later.
Clinical & Laboratory Features
| Feature | Classic Males | Heterozygous Females | Late-Onset Variants |
|---|---|---|---|
| Microalbuminuria onset | ~10–15 years | ~20–40 years | ~40–60 years |
| Overt proteinuria | ~20–30 years | Variable | Variable |
| eGFR decline rate | 5–7 mL/min/year | 1–3 mL/min/year | 2–5 mL/min/year |
| ESKD risk (cumulative) | ~50% by age 45 | ~15–20% by age 60 | Renal variant: high |
| Hypertension | Common after age 30 | Age-related | Common |
| Haematuria | Rare | Rare | Rare |
Renal Biopsy Findings
Renal biopsy may be performed when Fabry nephropathy is suspected but the diagnosis is unconfirmed, or to assess disease burden and guide treatment initiation. Key histological findings include:
- Light microscopy: Vacuolated podocytes (on semithin sections), focal segmental glomerulosclerosis, arteriolar hyalinosis, and varying degrees of tubulointerstitial fibrosis.
- Electron microscopy: Characteristic concentric lamellar ("myelin-like" or "zebra body") inclusions within lysosomes of podocytes, mesangial cells, endothelial cells, and tubular epithelial cells — pathognomonic.
- Immunofluorescence: Non-specific; may show IgM and C3 in sclerotic glomeruli.
Monitoring Renal Disease
Annual renal monitoring is recommended for all diagnosed Fabry patients:
- eGFR (CKD-EPI equation) — consider cystatin C–based eGFR for more accurate estimation in patients with disproportionate muscle mass changes.
- Urine albumin-to-creatinine ratio (uACR) — spot morning sample; grade as A1 (<3 mg/mmol), A2 (3–30), A3 (>30).
- Blood pressure — target <130/80 mmHg per KDIGO and RACGP guidelines.
- Renal ultrasound — assess kidney size; kidneys may be normal or slightly enlarged in early disease.
- Plasma lyso-Gb3 — serial monitoring to assess disease activity and treatment response.
Systemic Features (Heart, Nervous System, Skin)
Cardiac Manifestations
Cardiac involvement is a major driver of morbidity and mortality in Fabry disease, and is the leading cause of death in heterozygous females.
- Left ventricular hypertrophy (LVH): Progressive concentric LVH develops due to Gb3 deposition in cardiomyocytes and subsequent fibrosis. Onset typically occurs from age 20–30 in classically affected males and 40–50 in females. Echocardiography may show a "binary sign" of endocardial hyperechogenicity in early disease. Cardiac MRI with late gadolinium enhancement (LGE) identifies areas of replacement fibrosis — a marker of irreversible myocardial damage and a predictor of arrhythmia and heart failure.
- Arrhythmias: Atrial fibrillation, supraventricular tachycardia, bradycardia (sick sinus syndrome), and ventricular tachycardia are common. Ambulatory ECG monitoring (24–72 hour Holter) is recommended annually. Consider implantable loop recorder for patients with cryptogenic syncope.
- Valvular disease: Mild mitral and aortic regurgitation may occur; significant valvular disease is uncommon.
- Accelerated coronary artery disease: Endothelial Gb3 deposition contributes to premature atherosclerosis. Aggressive cardiovascular risk factor management is essential.
- Heart failure: Both heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF) may develop. Standard heart failure pharmacotherapy (ACEi/ARB, beta-blocker, mineralocorticoid receptor antagonist, SGLT2 inhibitor) should be used.
Cerebrovascular & Neurological Manifestations
- Stroke & TIA: Ischaemic stroke occurs at a mean age of approximately 33 years in classically affected males — far younger than the general population. Posterior circulation strokes are disproportionately common due to vertebrobasilar dolichoectasia. Haemorrhagic stroke may also occur.
- White matter lesions: MRI FLAIR hyperintensities are present in the majority of adult patients and may be seen in adolescents. These lesions are thought to reflect small vessel disease from endothelial Gb3 accumulation.
- Vertebrobasilar dolichoectasia: Dilatation and tortuosity of the basilar artery is a characteristic finding on MRA, present in up to 30% of patients.
- Peripheral neuropathy: Small-fibre neuropathy causes the hallmark acroparaesthesia (burning, lancinating pain in hands and feet), which typically begins in early childhood and may be misdiagnosed as erythromelalgia, rheumatic fever, or growing pains. Large-fibre neuropathy is less common.
- Autonomic dysfunction: Hypohidrosis or anhidrosis, gastrointestinal dysmotility (diarrhoea, abdominal cramping, early satiety), orthostatic hypotension, and impaired lacrimation.
- Hearing loss & tinnitus: Sensorineural hearing loss occurs in approximately 40% of patients, typically high-frequency.
Dermatological Features
- Angiokeratomas: Clusters of dark red-to-purple, non-blanching, slightly raised papules (1–3 mm) distributed in a "bathing trunk" pattern — buttocks, groin, periumbilical area, and upper thighs. They appear in adolescence and increase in number with age. Histologically, they represent ectatic dermal capillaries covered by hyperkeratotic epidermis.
- Hypohidrosis / Anhidrosis: Present in >80% of classically affected males by adolescence; leads to heat intolerance, exercise intolerance, and impaired thermoregulation.
- Lymphoedema: Peripheral lymphoedema of the lower limbs may develop due to Gb3 deposition in lymphatic endothelium.
Ophthalmological Features
- Corneal verticillata (vortex keratopathy): Whorl-like, golden-brown opacities in the corneal epithelium visible on slit-lamp examination. Present in virtually all classically affected males and most heterozygous females. These are pathognomonic, non-progressive, and do not affect vision. Notably, they are also caused by amiodarone and chloroquine — drug history is essential.
- Posterior spoke-like cataracts: Lens opacities that may appear in adolescence.
- Tortuous retinal & conjunctival vessels: A subtle but characteristic finding.
Gastrointestinal Features
Gastrointestinal symptoms affect 30–70% of patients and significantly impair quality of life. Diarrhoea (often postprandial), abdominal cramping, nausea, early satiety, and constipation are attributed to autonomic dysfunction and Gb3 deposition in mesenteric vasculature and neural plexuses. Dietary modification (small, frequent, low-fat meals) and symptomatic pharmacotherapy (loperamide, domperidone) form the mainstay of management.
Investigations
The diagnostic workup for Fabry disease involves biochemical, genetic, and organ-specific assessments. In Australia, these investigations are available through specialised laboratories and tertiary centres.
Risk Stratification & Severity Scoring
Risk stratification in Fabry disease informs the urgency and intensity of treatment. The Mainz Severity Score Index (MSSI) and the Fabry-specific tools developed by the European Fabry Working Group provide a framework for clinical staging.
Management (Enzyme Replacement Therapy)
Enzyme Replacement Therapy (ERT)
ERT has been the cornerstone of Fabry disease treatment since 2001. Two recombinant enzyme preparations are available in Australia, both PBS-listed under Section 100 (Special Authority) arrangements.
PBS Authority Criteria for ERT
Under the Pharmaceutical Benefits Scheme, ERT with agalsidase alfa or agalsidase beta is available as a Section 100 (Special Authority) benefit for patients meeting ALL of the following:
- Confirmed diagnosis of Fabry disease by α-Gal A enzyme assay and/or GLA gene sequencing.
- Documented evidence of clinical disease activity — either symptomatic (neuropathic pain, angiokeratoma, hypohidrosis) AND/OR evidence of end-organ involvement (proteinuria, reduced eGFR, LVH, cerebral white matter lesions).
- Prescribed by or in consultation with a specialist experienced in the management of Fabry disease (e.g., metabolic physician, nephrologist, or clinical geneticist).
- Continued access requires demonstrated clinical stability or improvement at 12-month review.
Pharmacological Chaperone Therapy — Migalastat
Migalastat (Galafold®) is an oral small-molecule pharmacological chaperone that binds to and stabilises specific misfolded α-Gal A mutant enzymes, facilitating trafficking from the endoplasmic reticulum to lysosomes where the enzyme can function.
Adjunctive & Supportive Management
- Renoprotective therapy: ACE inhibitor (e.g., perindopril 5–10 mg daily) or ARB (e.g., irbesartan 150–300 mg daily) titrated to maximum tolerated dose. Target uACR <3 mg/mmol; if not achieved, consider dual blockade with caution (monitor K⁺ and creatinine). SGLT2 inhibitors (dapagliflozin 10 mg daily, empagliflozin 10 mg daily) — PBS-listed for CKD; emerging data support use in Fabry nephropathy; monitor for euglycaemic ketoacidosis.
- Antihypertensive therapy: Target BP <130/80 mmHg. ACEi/ARB preferred as first-line (dual renoprotective role).
- Neuropathic pain: First-line: pregabalin 75–300 mg BD or gabapentin 300–1200 mg TDS (both PBS-listed). Alternatives: carbamazepine 200–400 mg BD (PBS-listed), duloxetine 60 mg daily. Avoid opioids for chronic neuropathic pain. Paracetamol and NSAIDs have limited efficacy.
- Cardiac management: Standard heart failure pharmacotherapy (ACEi/ARB, beta-blocker, MRA, SGLT2i) as per ESC/NHF guidelines. Anticoagulation for AF (CHA₂DS₂-VASc ≥2 in males, ≥3 in females). Consider ICD for sustained VT or high-risk features. Pacemaker for symptomatic bradycardia.
- Cerebrovascular risk reduction: Antiplatelet therapy (aspirin 100 mg daily) for secondary stroke prevention; primary prevention in high-risk patients per clinician judgement. Aggressive management of modifiable risk factors (hypertension, dyslipidaemia, smoking cessation).
- GI symptom management: Dietary modification (small, frequent, low-fat meals). Loperamide 2–4 mg PRN for diarrhoea. Domperidone 10 mg TDS for gastroparesis. Consider pancreatic enzyme supplementation if steatorrhoea present.
- Exercise & heat avoidance: Graduated exercise programme (avoiding extremes of heat). Occupational therapy for hypohidrosis (cooling vests, hydration strategies).
- Psychosocial support: Fabry disease significantly impacts quality of life. Referral to psychology/psychiatry for chronic pain management, adjustment disorder, and depression screening. Patient support groups (e.g., Fabry Australia, Australian Fabry Disease Patient Support Group).
Renal Transplantation
Kidney transplantation is an effective treatment for Fabry-related ESKD. Patient and graft survival rates are comparable to other causes of ESKD, though recurrence of Gb3 deposition in the allograft occurs over time. ERT should be continued post-transplant. Pre-transplant cardiac and cerebrovascular screening is essential. Combined heart–kidney transplantation may be considered in patients with concurrent severe cardiomyopathy.
Emerging Therapies
Gene therapy (adeno-associated virus [AAV]-mediated GLA gene delivery) is under active investigation in phase I/II clinical trials. Substrate reduction therapy with lucerastat (an inhibitor of glucosylceramide synthase) is in late-phase development. These therapies are not currently available outside clinical trials in Australia.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Germain DP. Fabry disease. Orphanet Journal of Rare Diseases. 2010;5:30. doi:10.1186/1750-1172-5-30.
- 2. Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney International. 2017;91(2):284–293.
- 3. Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Molecular Genetics and Metabolism. 2018;123(4):416–427.
- 4. Hopkin RJ, Jefferies JL, Laney DA, et al. The management and treatment of children with Fabry disease: A United States-based perspective. Molecular Genetics and Metabolism. 2016;117(2):104–113.
- 5. Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641–659.
- 6. Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: A multicenter study. Journal of the American Society of Nephrology. 2017;28(5):1631–1641.
- 7. Nicholls K, Becker G, Walker R, et al. Enzyme replacement therapy in Fabry disease. Internal Medicine Journal. 2008;38(10):770–778.
- 8. Hughes DA, Nicholls K, Shanmuganathan SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. Journal of Medical Genetics. 2017;54(4):288–296.
- 9. Australian Government Department of Health and Aged Care. Pharmaceutical Benefits Scheme: Agalsidase alfa, agalsidase beta, and migalastat. PBS Schedule. Available at: https://www.pbs.gov.au. Accessed 2024.
- 10. Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney International. 2024;105(4S):S117–S314.
- 11. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2020 summary report. Canberra: AIHW; 2020.
- 12. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genetics in Medicine. 2006;8(9):539–548.
- 13. Biegstraaten M, Arngrímsson R, Barbey F, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet Journal of Rare Diseases. 2015;10:36.
- 14. Wanner C, Arad M, Baron R, et al. European expert consensus statement on the diagnosis and management of Fabry disease. European Journal of Human Genetics. 2024;32:513–525.