📋 Key Information Summary
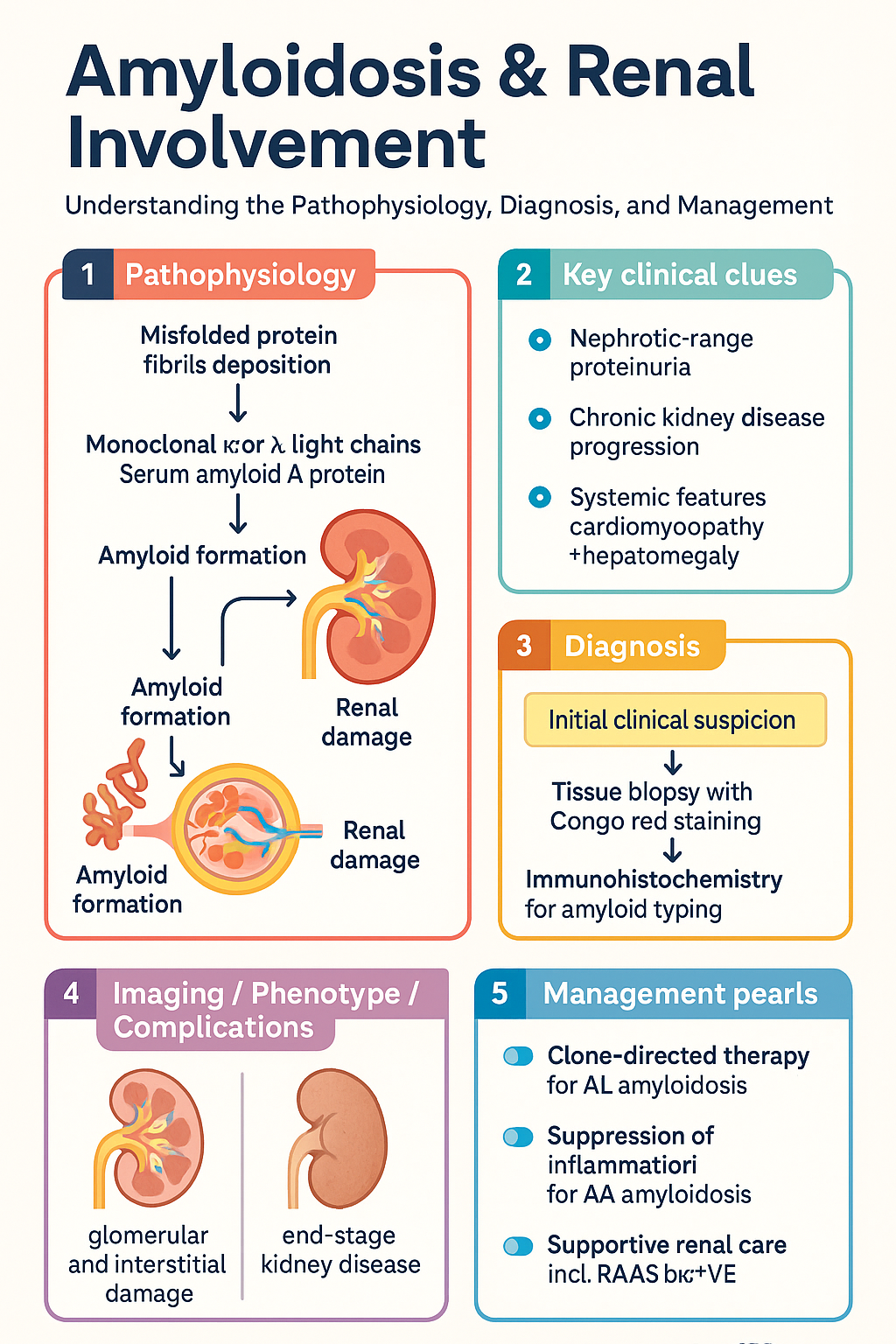
- Renal amyloidosis results from extracellular deposition of misfolded protein fibrils in the glomeruli and interstitium, most commonly AL (immunoglobulin light-chain) or AA (serum amyloid A) type.
- AL amyloidosis is the most frequent systemic amyloidosis in Australia; it arises from a clonal plasma-cell or B-cell disorder and deposits monoclonal κ or λ light chains.
- AA amyloidosis complicates chronic inflammatory conditions (rheumatoid arthritis, FMF, chronic infections) and deposits serum amyloid A fragments.
- Renal involvement presents predominantly with nephrotic-range proteinuria (≥3.5 g/day), progressing to chronic kidney disease and often end-stage kidney disease (ESKD).
- Diagnosis requires tissue biopsy with Congo red staining and apple-green birefringence under polarised light; renal biopsy has >95 % sensitivity for renal amyloid.
- Serum free light-chain (FLC) assay and immunofixation electrophoresis are essential to differentiate AL from AA type and to monitor treatment response.
- Technetium-99m-labelled SAP scintigraphy (where available) quantifies whole-body amyloid load but does not substitute for biopsy.
- AL amyloidosis treatment targets the underlying clone: bortezomib-based regimens (VCD) are first-line in Australia, with autologous stem-cell transplant in selected patients.
- AA amyloidosis management centres on suppressing the underlying inflammatory disease (biologic DMARDs, colchicine for FMF) to reduce SAA production to <4 mg/L.
- Eplagersen (an anti-siRNA targeting TTR) is PBS-listed for hereditary ATTR amyloidosis; AL amyloidosis therapies are funded via hospital authority and clinical trials.
- Aboriginal and Torres Strait Islander peoples have higher rates of chronic infections (rheumatic heart disease, bronchiectasis) predisposing to AA amyloidosis.
- Nephrology referral is mandatory for all suspected renal amyloidosis; haematology/oncology co-management is essential for AL amyloidosis.
- Supportive renal care includes RAAS blockade for proteinuria, loop diuretics for oedema, statin therapy, and VTE prophylaxis for severe nephrotic syndrome.
Introduction & Australian Epidemiology
The amyloidoses are a group of disorders characterised by extracellular deposition of insoluble fibrillar proteins derived from normally soluble precursors. These fibrils adopt a cross-β-sheet configuration, bind Congo red dye, and exhibit apple-green birefringence under polarised light microscopy. When fibrils accumulate in the kidney, they cause progressive glomerular and interstitial damage, manifesting as nephrotic-range proteinuria and chronic kidney disease (CKD).
Of the >30 known amyloidogenic proteins, two types account for the vast majority of renal amyloidosis encountered in Australian clinical practice:
- AL (primary) amyloidosis — derived from monoclonal immunoglobulin light chains produced by a clonal plasma-cell or B-cell neoplasm.
- AA (secondary) amyloidosis — derived from serum amyloid A (SAA), an acute-phase reactant produced during chronic inflammation.
Australian epidemiology: AL amyloidosis is the most common systemic amyloidosis diagnosed in Australia, with an estimated incidence of 8–12 per million per year. The median age at diagnosis is 63–65 years. Renal involvement occurs in approximately 50–70 % of AL cases. AA amyloidosis is rarer overall but remains clinically important in populations with chronic inflammatory diseases, particularly Aboriginal and Torres Strait Islander communities where rheumatic heart disease and suppurative lung disease persist at high prevalence. The Australian Amyloidosis Network (AAN) and the Australian and New Zealand Society of Nephrology (ANZSN) maintain disease registries that inform practice.
AL vs AA Amyloidosis in the Kidney
Distinguishing AL from AA amyloidosis is critical because their treatments differ fundamentally. AL amyloidosis requires clone-directed therapy (chemotherapy), whereas AA amyloidosis requires suppression of the inflammatory driver.
| Feature | AL Amyloidosis | AA Amyloidosis |
|---|---|---|
| Precursor protein | Monoclonal κ or λ immunoglobulin light chain | Serum amyloid A (SAA) protein |
| Underlying cause | Clonal plasma-cell or B-cell disorder (MGUS, myeloma) | Chronic inflammation — RA, FMF, IBD, bronchiectasis, chronic osteomyelitis |
| Typical age | 60–70 years | Variable; younger in FMF |
| Renal presentation | Nephrotic syndrome, rapid CKD progression | Nephrotic syndrome, slower CKD progression |
| Extrarenal organs | Heart (>70 %), liver, peripheral nerves, soft tissues, GI tract | Spleen, liver, GI tract; heart involvement uncommon |
| Serum FLC | Abnormal κ/λ ratio in >90 % | Normal FLC ratio |
| Immunohistochemistry | Positive for κ or λ light chain | Positive for SAA; negative for light chains |
| First-line treatment | Bortezomib-based chemotherapy (± ASCT) | Treat underlying inflammation (biologics, colchicine) |
| Median renal survival (untreated) | 12–18 months to ESKD | 3–7 years to ESKD |
Pathophysiology
In AL amyloidosis, monoclonal light chains—most commonly λ6—are secreted by a low-grade clonal plasma-cell population. These light chains misfold, resist proteolysis, and deposit as fibrils predominantly in the mesangium, glomerular basement membrane, and peritubular capillaries. Fibril deposition disrupts the glomerular filtration barrier, causing heavy proteinuria and podocyte dysfunction.
In AA amyloidosis, persistently elevated SAA (produced by hepatocytes under IL-6, IL-1, and TNF-α stimulation) is cleaved by macrophage enzymes into amyloid fibrils. Deposition is predominantly glomerular but extends to the medulla and vasculature. The degree of renal AA amyloidosis correlates with the magnitude and duration of SAA elevation.
Clinical Features (Nephrotic Syndrome, CKD)
Renal amyloidosis may be asymptomatic at early stages, detected incidentally by proteinuria on urinalysis. As disease progresses, characteristic clinical syndromes emerge.
Nephrotic Syndrome
- Heavy proteinuria (≥3.5 g/day; frequently >5 g/day)
- Hypoalbuminaemia (serum albumin <25 g/L)
- Generalised oedema (peripheral, periorbital, ascites, pleural effusion)
- Hypercholesterolaemia and hypertriglyceridaemia
- Foamy or frothy urine
Chronic Kidney Disease
Progressive decline in eGFR is characteristic. In AL amyloidosis, CKD may progress rapidly to ESKD within 12–24 months if the underlying clone is untreated. In AA amyloidosis, the trajectory is more indolent but relentless if inflammation persists.
Extrarenal Features — AL Amyloidosis
Clues to systemic AL amyloidosis include:
- Cardiac: Restrictive cardiomyopathy, low-voltage ECG with pseudo-infarct pattern, raised NT-proBNP and troponin
- Hepatic: Hepatomegaly with elevated ALP, occasionally cholestatic jaundice
- Peripheral neuropathy: Symmetric sensory-predominant, autonomic (orthostatic hypotension, gastroparesis)
- Soft-tissue: Macroglossia, periorbital purpura (pathognomonic), carpal tunnel syndrome
- Gastrointestinal: Dysmotility, malabsorption, pseudo-obstruction, GI bleeding
Extrarenal Features — AA Amyloidosis
- Splenomegaly (common, may cause hyposplenism)
- Hepatomegaly
- Adrenal gland involvement (rare clinical insufficiency)
- GI tract involvement (usually subclinical)
Investigations (Biopsy, SAP Scan, Serum FLC)
A systematic investigation pathway is essential to confirm amyloidosis, determine the amyloid type, and assess organ involvement.
Investigation Algorithm
Management (Treat Underlying Disease)
The overarching principle of amyloidosis management is to reduce production of the amyloidogenic precursor protein. In AL amyloidosis this means eliminating the clonal plasma-cell population; in AA amyloidosis it means suppressing the underlying chronic inflammation. Supportive renal care is provided in parallel.
AL Amyloidosis — Clone-Directed Therapy
Treatment should be initiated promptly after diagnosis in conjunction with a haematologist experienced in amyloidosis (via state amyloidosis centres of excellence).
AL Amyloidosis — Response Criteria
| Response Level | Haematologic Criteria | Organ Response |
|---|---|---|
| Complete response (CR) | Negative serum and urine immunofixation; normal FLC ratio | ≥30 % reduction in 24-hr proteinuria if nephrotic |
| Very good partial response (VGPR) | dFLC <40 mg/L | eGFR improvement or stabilisation |
| Partial response (PR) | ≥50 % reduction in dFLC | May take 6–12 months to manifest |
| No response / progression | <50 % reduction or rising dFLC | Worsening proteinuria, rising creatinine |
AA Amyloidosis — Suppress the Inflammatory Driver
The primary therapeutic target is to maintain serum SAA <4 mg/L. This requires aggressive management of the underlying inflammatory or infectious disease.
Supportive Renal Care (AL and AA)
While disease-modifying therapy targets the precursor protein, supportive measures reduce symptoms and slow CKD progression.
Risk Stratification / Severity Scoring
AL amyloidosis staging determines prognosis and guides treatment intensity. The Revised Mayo 2012 staging system is most widely used:
Renal Risk Stratification (AL Amyloidosis)
Renal staging (Palladini criteria):
- Renal stage I: Proteinuria <5 g/day AND eGFR ≥50 mL/min
- Renal stage II: Either proteinuria ≥5 g/day OR eGFR <50 mL/min
- Renal stage III: Proteinuria ≥5 g/day AND eGFR <50 mL/min
Median time to ESKD: Stage I >10 years; Stage II ~4 years; Stage III ~1.5 years.
Monitoring
Regular monitoring assesses treatment response, disease progression, and organ function.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on difference between involved and uninvolved free light chain levels. Haematologica. 2012;97(12):1819–1824.
- 2. Kastritis E, Palladini G, Minnema MC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385(1):46–58. (ANDROMEDA trial)
- 3. Palladini G, Hegenbart U, Milani P, et al. A staging system for renal outcome and an early warning system for cardiac risk in AL amyloidosis. Blood. 2014;123(15):2310–2317.
- 4. Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–995.
- 5. Lachmann HJ, Goodman HJ, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361–2371.
- 6. Wechalekar AD, Gillmore JD, Bird J, et al. Guidelines on the management of AL amyloidosis. Br J Haematol. 2015;168(2):186–206.
- 7. Gillmore JD, Gane E, Taubel J, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21.
- 8. Australian Institute of Health and Welfare (AIHW). Chronic kidney disease. Cat. no. PHE 229. Canberra: AIHW; 2023.
- 9. RHDAustralia (ARF/RHD writing group). The 2020 Australian guideline for prevention, diagnosis and management of acute rheumatic fever and rheumatic heart disease. 3rd ed. Darwin: Menzies School of Health Research; 2020.
- 10. Cheung RC, Nandakumar M, Craig JC. The burden of chronic kidney disease in Indigenous Australians: a systematic review. Intern Med J. 2010;40(3):193–199.
- 11. Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751–3757.
- 12. Cibeira MT, Sanchorawala V, Seldin DC, et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood. 2011;118(16):4346–4352.
- 13. Holmgren G, Ericzon BG, Groth CG, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113–1116.
- 14. Australian Commission on Safety and Quality in Health Care (ACSQHC). National Safety and Quality Health Service Standards. 2nd ed. Sydney: ACSQHC; 2021.